

- •Preface
- •Acknowledgments
- •Contents
- •Contributors
- •1. Introduction
- •2. Evaluation of the Craniomaxillofacial Deformity Patient
- •3. Craniofacial Deformities: Review of Etiologies, Distribution, and Their Classification
- •4. Etiology of Skeletal Malocclusion
- •5. Etiology, Distribution, and Classification of Craniomaxillofacial Deformities: Traumatic Defects
- •6. Etiology, Distribution, and Classification of Craniomaxillofacial Deformities: Review of Nasal Deformities
- •7. Review of Benign Tumors of the Maxillofacial Region and Considerations for Bone Invasion
- •8. Oral Malignancies: Etiology, Distribution, and Basic Treatment Considerations
- •9. Craniomaxillofacial Bone Infections: Etiologies, Distributions, and Associated Defects
- •11. Craniomaxillofacial Bone Healing, Biomechanics, and Rigid Internal Fixation
- •12. Metal for Craniomaxillofacial Internal Fixation Implants and Its Physiological Implications
- •13. Bioresorbable Materials for Bone Fixation: Review of Biological Concepts and Mechanical Aspects
- •14. Advanced Bone Healing Concepts in Craniomaxillofacial Reconstructive and Corrective Bone Surgery
- •15. The ITI Dental Implant System
- •16. Localized Ridge Augmentation Using Guided Bone Regeneration in Deficient Implant Sites
- •17. The ITI Dental Implant System in Maxillofacial Applications
- •18. Maxillary Sinus Grafting and Osseointegration Surgery
- •19. Computerized Tomography and Its Use for Craniomaxillofacial Dental Implantology
- •20B. Atlas of Cases
- •21A. Prosthodontic Considerations in Dental Implant Restoration
- •21B. Overdenture Case Reports
- •22. AO/ASIF Mandibular Hardware
- •23. Aesthetic Considerations in Reconstructive and Corrective Craniomaxillofacial Bone Surgery
- •24. Considerations for Reconstruction of the Head and Neck Oncologic Patient
- •25. Autogenous Bone Grafts in Maxillofacial Reconstruction
- •26. Current Practice and Future Trends in Craniomaxillofacial Reconstructive and Corrective Microvascular Bone Surgery
- •27. Considerations in the Fixation of Bone Grafts for the Reconstruction of Mandibular Continuity Defects
- •28. Indications and Technical Considerations of Different Fibula Grafts
- •29. Soft Tissue Flaps for Coverage of Craniomaxillofacial Osseous Continuity Defects with or Without Bone Graft and Rigid Fixation
- •30. Mandibular Condyle Reconstruction with Free Costochondral Grafting
- •31. Microsurgical Reconstruction of Large Defects of the Maxilla, Midface, and Cranial Base
- •32. Condylar Prosthesis for the Replacement of the Mandibular Condyle
- •33. Problems Related to Mandibular Condylar Prosthesis
- •34. Reconstruction of Defects of the Mandibular Angle
- •35. Mandibular Body Reconstruction
- •36. Marginal Mandibulectomy
- •37. Reconstruction of Extensive Anterior Defects of the Mandible
- •38. Radiation Therapy and Considerations for Internal Fixation Devices
- •39. Management of Posttraumatic Osteomyelitis of the Mandible
- •40. Bilateral Maxillary Defects: THORP Plate Reconstruction with Removable Prosthesis
- •41. AO/ASIF Craniofacial Fixation System Hardware
- •43. Orbital Reconstruction
- •44. Nasal Reconstruction Using Bone Grafts and Rigid Internal Fixation
- •46. Orthognathic Examination
- •47. Considerations in Planning for Bimaxillary Surgery and the Implications of Rigid Internal Fixation
- •48. Reconstruction of Cleft Lip and Palate Osseous Defects and Deformities
- •49. Maxillary Osteotomies and Considerations for Rigid Internal Fixation
- •50. Mandibular Osteotomies and Considerations for Rigid Internal Fixation
- •51. Genioplasty Techniques and Considerations for Rigid Internal Fixation
- •52. Long-Term Stability of Maxillary and Mandibular Osteotomies with Rigid Internal Fixation
- •53. Le Fort II and Le Fort III Osteotomies for Midface Reconstruction and Considerations for Internal Fixation
- •54. Craniofacial Deformities: Introduction and Principles of Management
- •55. The Effects of Plate and Screw Fixation on the Growing Craniofacial Skeleton
- •56. Calvarial Bone Graft Harvesting Techniques: Considerations for Their Use with Rigid Fixation Techniques in the Craniomaxillofacial Region
- •57. Crouzon Syndrome: Basic Dysmorphology and Staging of Reconstruction
- •58. Hemifacial Microsomia
- •59. Orbital Hypertelorism: Surgical Management
- •60. Surgical Correction of the Apert Craniofacial Deformities
- •Index
54
Craniofacial Deformities: Introduction and Principles of Management
G.E. Ghali, Wichit Tharanon, and Douglas P. Sinn
In the last several years the arena of craniomaxillofacial surgery has expanded in scope, and the treatment of these deformities has become more sophisticated. In view of these many changes, the embryology, etiology, pathogenesis, imaging, and treatment of the common craniofacial deformities will be reviewed with their management.
Embryology of the Craniofacial Region
Any discussion of craniofacial syndromes must be preceded by a consideration of the normal embryologic development of these structures. The neural crest cells are important to the development of the facial skeleton. Translocated neural crest cells, upon reaching their destination, differentiate into cartilage, bone, and ligaments of the face and contribute to the muscles and arteries in the region. Any disruption of migration and differentiation may have deleterious effects.1 The crucial period of organogenesis takes place during the first 12 weeks of gestation, and it is during this time that the majority of congenital craniofacial deformities are established.1,2 The facial growth centers appear at the end of the third week of embryonic life and are in their definitive place by the eighth embryonic week. The face derives its morphology from five prominences. These prominences are the single frontonasal and the paired maxillary and mandibular processes. The grooves between these facial prominences usually disappear by the seventh week of gestation. A persisting groove will generally result in a congenital facial cleft. Occipital somites and somitomeres form a majority of the neurocranium. The neurocranium is anatomically divided into two portions: the membranous part consisting of flat bones, which surround the brain as a vault, and the cartilaginous part or chondrocranium, which forms the bones of the skull base. The sides and roof of the skull develop from mesenchyme, invest the brain, and eventually undergo membranous ossification. Membranous bones are characterized by the presence of needle-like spicules. These spicules progressively radiate from the primary ossification centers toward the periphery (Figure 54.1). Membranous bone enlarges by apposition of new layers on
the outer surface and by simultaneous osteoclastic resorption from the interior.
At birth, the flat bones of the skull are separated from each other by narrow seams of connective tissue, the cranial sutures. At points where more than two bones meet, the sutures are wide and known as fontanelles. The sutures and fontanelles allow the bones of the skull to overlap during the birth process. Several of the sutures and the fontanelles remain membranous for a considerable time after birth. Growth of the bones of the vault is the result of expansion of these flat bones caused mainly by the volumetric growth of the brain. Although a 5- to 7-year-old child has normally achieved maximum cranial capacity, some of the sutures remain open until adulthood. Cartilaginous neurocranium or chondrocranium consists initially of a number of separate cartilages. When these cartilages fuse and ossify by endochondral ossification, the base of the skull is formed.3
Etiology of Craniofacial Deformities
Congenital craniofacial deformities can have a genetic, environmental, or combined etiology. Currently, it is known that environmental factors associated with congenital deformities are radiation, infection, maternal idiosyncrasies, and chemical agents:4
Radiation. Large doses of radiation have been associated with microcephaly.
Infection. The children of mothers affected with toxoplasmosis, rubella, or cytomegalovirus show increased frequency of facial clefts.
Maternal idiosyncrasies. Numerous studies attest to the major role of maternal factors such as age, weight, and general health on the resistance or susceptibility of the developing embryo to potential causes of malformation.
Chemical. Many chemical agents or drugs have been implicated in craniofacial malformation such as ethanol, 13-cis retinoic acid, and methotrexate.5 Additionally, other drugs suspected of playing a role in craniofacial syndromes are the anticonvulsants.
671

672 |
G.E. Ghali, W. Tharanon, and D.P. Sinn |
FIGURE 54.1 Skull of infant demonstrating primary cranial ossification centers.
Principles of Management
of Craniofacial Deformities
Multidisciplinary Team Approach
The multidisciplinary team concept was developed from the recognition that failures commonly occurred when various aspects of care were not coordinated and when the relationships among coexisting problems were not known. The objectives of this approach are diagnosis, formulation, and execution of treatment plans as well as longitudinal follow-up for patients with craniofacial deformities; the team should meet at least monthly for regular outpatient evaluations. Transcripts of these evaluations are forwarded with treatment recommendations to primary care providers and appropriate agencies. Children under 5 years ofageareusuallyevaluatedannually,whereasthoseover5years of age are seen every other year. The frequency of evaluation varies with the stability of the deformity and its consequences. The craniofacial team should consist of an anesthesiologist, an ophthalmologist, a surgeon (plastic and/or oral/maxillofacial), an audiologist, a maxillofacial prosthodontist, an orthodontist, a psychologist, a geneticist, an otolaryngologist, a radiologist, a neurologist, a neurosurgeon, a pediatrician, social services, a pedodontist, a speech pathologist, an orthotist, and a nurse.6,7
Genetic Diagnosis: The Dysmorphology
Examination
Dysmorphology is the study of birth defects with an emphasis on understanding the mechanisms of morphogenesis. The
dysmorphologist or clinical geneticist should be one of the first clinicians to evaluate the patient with a birth defect involving the craniomaxillofacial region. This approach benefits both the patient and the team caring for the patient by fostering a comprehensive approach to evaluation, especially of defects outside the craniofacial region. Accurate diagnosis then becomes the basis for accurate prognosis and recurrence risk (genetic) counseling.
Genetic Counseling
Genetic counseling provides information and emotional support to families in which an individual has a disorder or birth defect. The genetic counselor gathers the family’s medical and pregnancy history, obtains necessary medical records, and determines the concerns and questions of the individuals attending the assessment. This service provides medical and psychosocial support to unaffected relatives as well as the affected individual. The genetic counselor may interact closely with other members of the craniofacial team by providing important information regarding the history, patient, and family.
Radiographic Evaluation
The radiographic evaluation of craniofacial deformities is used to quantitatively define aberrant anatomy, plan surgical procedures, and evaluate the effects of growth as well as surgery. Conventional skull radiographs such as plain skull films and lateral cephalograms are inexpensive and widely available. The preoperative assessment of patients with suspected or known craniofacial deformities is based on these conventional radiographs. The majority of synostosis can be demonstrated on plain skull films. Normal or patent cranial sutures manifest as a line and the absence of a radiolucent line in the normal anatomic position of a suture may suggest a craniosynostosis.
Currently, computer tomography (CT) scans provide improved hard tissue imaging.8–10 The definition of these elements of the bony facial structures on high-resolution CT images is unmatched by other imaging techniques (such as plain skull or tomogram). The development of CT scanning, particularly three-dimensional reformatting, and the maturation of readily available means of craniofacial surgery has led to a close dependence on CT scanning for preoperative surgical planning. Additionally, CT scanning has also been used to document surgical changes in vivo and to follow them longi- tudinally.11–24
Common Craniofacial Deformities
In general, craniofacial deformities can be divided into three major subgroups: those involving the cranial skeleton only (coronal, sagittal, metopic, and lambdoidal craniosynostosis), those involving the cranial and facial skeleton (Crouzon’s, Apert’s, and Pfeiffer’s syndromes), and those involving the facial skeleton only (Treacher Collins syndrome, hemifacial microsomia, cleft lip, and palate).
54. Craniofacial Deformities: Introduction and Principles of Management |
673 |
Craniosynostosis
In 1851, Virchow was credited as the first to use the term craniosynostosis in describing a disorder characterized by an abnormal shape of the skull. Virchow noted that synostosis in the skull restricted growth perpendicular to the direction of the suture and promoted compensatory overgrowth parallel to it.25–27
Pathogenesis of Craniosynostosis
The pathogenesis of craniosynostosis is complex and probably multifactorial. Moss theorized that abnormal tensile forces are transmitted to the dura from an anomalous cranial base through key ligamentous attachments leading to craniosynostosis such as seen in Apert’s and Crouzon’s syndromes.28 This hypothesis fails to explain the coexistence of craniosynostosis in those patients with a normal cranial base configuration. The etiology of craniosynostosis may be postulated to be the result of either primary suture abnormalities, sufficient extrinsic forces that overcome the underlying expansive forces of the brain, or inadequate intrinsic growth forces of the brain.29–31
Limitation of Brain Growth
Brain volume in the normal child almost triples during the first year of life. By 2 years of age, the cranial capacity is four times that at birth. If the brain growth is to proceed unhindered, open sutures at the level of the cranial vault and base must spread during phases of rapid growth, resulting in marginal ossification.
In craniosynostosis, premature suture fusion is combined with continuing brain growth. Depending on the number and location of prematurely fused sutures and the timing of closure, the growth potential of the brain may be limited. Surgical intervention, with suture release and reshaping, is done to restore a more normal intracranial volume. In general, this does not reverse the process, and diminished volume is often the end result.35
Neuropsychiatric Disorders
Neuropsychiatric disorders are thought to be secondary to cerebral compression and range from mild behavioral disturbances to overt mental retardation. Several studies have shown that children with craniosynostosis and associated neuropsychiatric disorders often improve after surgery.36–39
Functional Problems Associated with Craniosynostosis
The major functional problems with craniosynostosis are intracranial hypertension, visual impairment, limitation of brain growth, and neuropsychiatric disorders.
Intracranial Hypertension
The clinical symptoms include headaches, irritability, and difficulty sleeping. The radiographic signs may include cortical thinning or a Lückenschadel (beaten metal) appearance of the inner table of the skull; these clinical and radiographic signs are relatively late developments. If intracranial hypertension goes untreated, it affects brain function; if persistent this may necessitate early operative intervention during the first few months of life. Intracranial hypertension most likely affects those with the greatest disparity between brain growth and intracranial capacity. Currently, intracranial volume is measured by using CT scans.32–34 This noninvasive method is used to measure intracranial volume in children with craniosynostosis. It might then be possible to select those individuals who are at a greater risk for developing intracranial hypertension and would benefit the most from early surgery.
Visual Impairment
Intracranial hypertension, if left untreated, leads to papilledema. Eventually, optic atrophy develops, resulting in partial or complete blindness. Some forms of craniosynostosis may involve orbital hypertelorism and may lead to compromised visual activity and restricted binocular vision.
Current Surgical Approach: Staging
of Reconstruction
In most cases, craniosynostosis suture release and cranial vault and orbital reshaping are mandatory before the child reaches 36 months of age.40–53 An intracranial approach is used for cranial vault and orbital osteotomies, with reshaping and advancement of bony segments for ideal age-appropriate bony morphology. When planning the timing and type of surgical intervention, one must take into account the functions, future growth, and development of the craniofacial skeleton, as well as the maintenance of a normal body image.35,54–57 In severe forms of craniosynostosis, additional revision of the cranial vault and orbit is necessary during infancy or early childhood to further increase intracranial volume; this allows for continued brain growth and avoids or reduces intracranial hypertension.58
Although many of the following examples depict transosseous wiring and/or titanium plating, our current trend is the utilization of resorbable plates and screws (Lactosorb, Walter Lorenz Surgical, Inc., Jacksonville, FL). These plates, composed of polylactic and polyglycolic acid, are completely resorbed by hydrolysis within 9 to 14 months while maintaining tensile strength for initial early stabilization. As a result, growth restrictions are minimized as is the potential for transcranial migration.
Classification of Craniosynostosis
The classification of craniosynostosis is based on the shape of the skull, which usually reflects the underlying prematurely fused suture(s).59–62 The major cranial vault sutures that may be involved include the left and/or right coronal, metopic, sagittal, and left and right lambdoid (Figure 54.2).

674 |
G.E. Ghali, W. Tharanon, and D.P. Sinn |
FIGURE 54.2 Three views depicting the major cranial sutures.
FIGURE 54.3 Anterior plagiocephaly illustrated from anterior and superior views.
a |
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.4 (a) Anterior view of child demonstrating plagiocephaly. (b) Lateral view of child demonstrating plagiocephaly.
54. Craniofacial Deformities: Introduction and Principles of Management |
675 |
Unilateral Coronal Synostosis
Unilateral coronal synostosis results in flatness or obliquity on the ipsilateral side of the forehead and supraorbital ridge region. The term for this deformity is anterior plagiocephaly (Figure 54.3). There are characteristic morphologic features on the affected ipsilateral side. The frontal bone is flat and the supraorbital ridge and lateral orbital rim are recessed. The affected orbit is shallow and the anterior cranial base is short in the anteroposterior (AP) dimension. The root of the nose may be constricted and deviated to the affected side. The ipsilateral zygoma and infraorbital rim may also be flat and recessed (Figure 54.4).
Timing and Surgical Management
Multiple surgical approaches for the correction of unilateral coronal synostosis have been described.40–44 Good long-term results are obtainable when treatment of unilateral coronal synostosis includes suture release and cranial vault orbital osteotomies with reshaping and advancement in infancy. At Children’s Medical Center at Dallas (CMC), unilateral orbital rim advancement (ORA) and frontal bone reshaping are ideally performed when the patient is 2.5 to 3 years of age. Other centers have reported good results when treated before 1 year of age. To achieve optimal symmetry, it is often necessary to use a bilateral surgical approach. Symmetry of the cranial vault and orbit must be achieved in surgery, since results generally do not improve over time. Stabilization is achieved by direct transosseous wires or resorbable plates and screws (Figure 54.5).
Bilateral Coronal Synostosis
This is the common cranial vault suture synostosis pattern associated with Apert and Crouzon’s syndromes. Bilateral coronal synostosis results in recession of the supraorbital ridge causing the overlying eyebrows to sit posterior to the corneas. The term for this cranial vault deformity is brachycephaly (Figure 54.6). The anterior cranial base is short in the AP dimension and wide transversely. The overlying cranial vault is high in the superior–inferior dimension, with anterior bulging of the upper forehead resulting from compensatory growth of the opening metopic suture. The orbits are often shallow (exorbitism), with the eyes bulging (exophthalmus) and abnormally separated (orbital hypertelorism).
Timing and Surgical Management
Treatment requires suture release and simultaneous bilateral orbital rim and frontal bone advancements. Surgery is performed when the patient is 2.5 to 3 years of age. Other centers have reported good results when treated before 1 year of age. The osteotomy for bilateral orbital rim advancement is made superior to the nasofrontal and frontozygomatic sutures and is extended to the squamous portion of the temporal bone. Stabilization is achieved with direct transosseous wires or
plates and screws (titanium or resorbable). The more normalized shape provides the needed increase in intracranial volume within the anterior cranial vault.
Metopic Synostosis
Metopic synostosis often occurs in isolation resulting in a triangular head or trigonocephaly (Figure 54.7). The associated cranial vault deformity consists of hypotelorism, an elevated supraorbital ridge medially and posteriorly, and inferior recession of the lateral orbital rims and lateral aspect of the supraorbital ridges. The bitemporal bony width is decreased, resulting in inappropriate anterior cranial vault shape and decreased anterior cranial vault volume. The overlying forehead is sloped posteriorly to about the level of the coronal sutures (Figure 54.8).
Timing and Surgical Management
Surgical treatment requires metopic suture release, simultaneous bilateral orbital rim advancements, and widening via a frontal bone advancement. These procedures are usually performed when the patient is 6 months to 1 year of age. Orbital hypotelorism is corrected by splitting the supraorbital ridge unit vertically in the midline and placing autogenous cranial bone grafts to increase the interorbital distance (Figure 54.9a). Stabilization is achieved with direct transosseous wires or resorbable microplate fixation. The microplate fixation is usually placed at the inner surface of the cranial bone (Figure 54.9b). The abnormally shaped forehead bone that has been removed is cut into sections of appropriate shape for the new forehead configuration. The anterior cranial base, anterior cranial vault, and orbits are given a more aesthetic shape, and the volume of the anterior cranial vault is increased allowing the brain adequate space. Autogenous bone may be taken from the posterior cranium, when required, to enhance frontal reconstruction.
Sagittal Suture Synostosis
Sagittal suture synostosis, the most common form of cranial vault synostosis, is rarely associated with increased intracranial pressure. The term for this cranial vault deformity is scaphocephaly (Figure 54.10). The deformity consists of an elongated anteroposterior dimension and a narrowed transverse dimension to the cranial vault. Usually, the midface and anterior cranial vault sutures are not affected.
Timing and Surgical Management
When premature closure of a sagittal suture is recognized early in infancy, most neurosurgeons believe that simple release of the sagittal suture through a strip craniectomy without simultaneous skull reshaping is adequate treatment.63 However, the residual cranial vault deformity may cause a continued psychosocial concern. If improvements in cranial

676 |
|
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a |
|
|
|
|
a |
b |
a |
b |
a |
b |
|
b
c
FIGURE 54.5 (a) Preoperative and postoperative superior views depicting a unilateral orbital rim advancement (ORA). (b) Three-quarters view depicting a unilateral ORA. (c) Intraoperative superior view. In this case, only unilateral reshaping was necessary to achieve symmetry.
vault shape are desired after 1 year of age, a formal total cranial vault reshaping is required (Figure 54.11).
Unilateral Lambdoid Synostosis
Unilateral lambdoid synostosis results in flatness of the affected ipsilateral parieto-occipital region. The location of the ear canal and external ear are more anterior on the ipsilateral side than on the contralateral side. This is more noticeable when the pa-
tient is examined from the superior view and relatively inconspicuous when observed from the frontal or profile view.
Timing and Surgical Management
Many surgeons consider either simple strip craniectomy of the involved suture or partial craniectomy of the region to be adequate treatment. We believe that a more extensive vault craniectomy and reshaping is generally necessary. If improve-
54. Craniofacial Deformities: Introduction and Principles of Management |
677 |
FIGURE 54.6 Brachycephaly illustrated from anterior and superior views.
FIGURE 54.7 Trigonocephaly illustrated from anterior and superior views.
a |
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.8 (a) Anterior view of child demonstrating subtle characteristics of metopic synostosis. (b) Lateral view of child demonstrating sloping of anterior forehead characteristic of trigonocephaly.

678 |
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a
b
FIGURE 54.9 (a) Correction of metopic synostosis as illustrated from lateral view. (b) Three-quarter view illustrating correction of metopic synostosis and position of microplate fixation.
ments in cranial vault shape are desired after 10 to 12 months of age, formal posterior cranial vault reshaping is often required.
third or fourth year. Occasionally, the synostosis may be evident at birth; rarely, no sutural involvement is noted.67 Increased intracranial pressure is frequent; therefore, it is mandatory to monitor the affected child.
Craniofacial Dysostosis
Crouzon Syndrome
This syndrome was first reported in 1912 by M.O. Crouzon. He described the characteristics of this syndrome: exorbitism, retromaxillism, inframaxillism, and paradoxical retrogenia.64 Inheritance is autosomal dominant and occurrence is both sporadic and familial. This condition affects about 1 in every 25,000 of the general population. Clinical appearance is characterized by recession of the frontal bone and supraorbital rim, retrusion of the midface, exorbitism with proptosis, and hypoplasia of the infraorbital rims (Figures 54.12a,b). Hypoplasia of the midface and a class III malocclusion is usually noted, but the mandible has normal growth potential. The skull is generally brachycephalic as a result of bilateral premature fusion of the coronal sutures.65,66 The synostosis commonly begins during the first year of life and is usually complete by the
Timing and Surgical Management
A staged approach is recommended for reconstruction in patients with Crouzon’s syndrome. In infancy, it is necessary to combinesuturereleasewithcranialvaultandorbitalosteotomies in addition to reshaping and advancement to correct the brachycephalic morphology and increase the intracranial volume (Figures 54.12c–h). If the intracranial pressure is increased, repeat craniotomy with further cranial vault and orbital shaping and advancement is required later in infancy or early childhood. The residual midface deficiency requires a LeFort III osteotomy with advancement when the patient is 5 to 7 years of age. This procedure may be combined with cranial vault reshaping to further increase the intracranial volume and relieve intracranial pressure or for improvements in cranial vault morphology. When skeletal maturity has been reached (14 to 16 years in females; 16 to 18 years in males), orthognathic surgery is indicated. A

54. Craniofacial Deformities: Introduction and Principles of Management |
679 |
FIGURE 54.10 Scaphocephaly illustrated from the lateral and superior views.
maxillary Le Fort I osteotomy and a genioplasty is generally needed for correction of any residual dentofacial deformities.68,69 Frequently, mandibular osteotomies will also be required if the anteroposterior discrepancy is great.
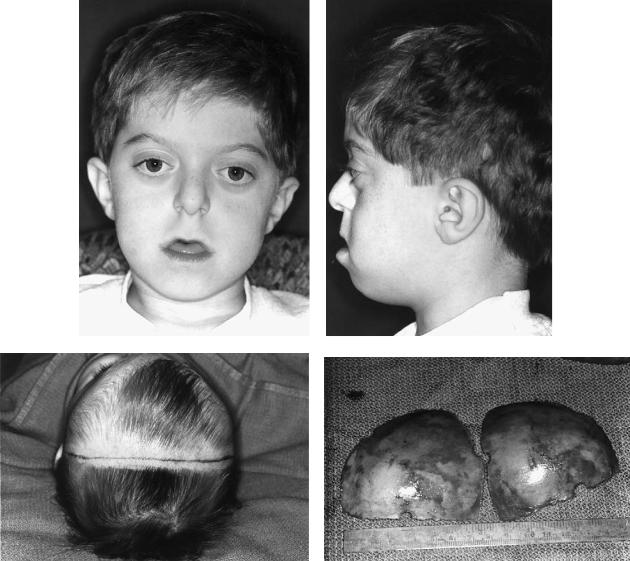
Apert Syndrome
Although Wheaton was the first to describe this syndrome in 1894, it is named after Apert, who in 1906 summarized four cases.70 He described the syndrome as a severe cranial vault deformity with associated syndactylism, with or without mental retardation and blindness. The incidence is reported to be 1:100,000 to 1:160,000 births.71 Although occurrence is sporadic, transmission is autosomal dominant. The clinical appearance is characterized by a flat face with hypertelorism, strabismus, and ocular muscle palsies, antimongoloid slant to the palpebral fissures and maxillary hypoplasia (Figure 54.13a).72,73 There is moderate to severe exorbitism, short zygomatic arches, and a prominent bregmatic bump. The fontanelles may be large and late in closing. The palate is narrow and either has a median groove or is clefted with a bifid uvula; the incidence of cleft palate approaches 30%. The limbs show bony syndactyly with complete fusion of the four fingers leaving the thumbs free (Figure 54.13b). The distal phalanx of the thumb is often broad. Cutaneous syndactyly of all toes may be either simple or complex (Figure 54.13c). Mental retardation is variably reported in association with Apert’s syndrome. The soft tissue drape is often abnormal and acne vulgaris with extensions to the forearm are common (70%). The facial skin, especially in the nasal region, is often thick
a |
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.11 Lateral views demonstrating (a) preoperative and (b) postoperative changes in anteroposterior morphology achieved with total vault reshaping.
680 |
|
|
|
|
|
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a |
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
c
d
FIGURE 54.12 (a) Anterior view of child demonstrating classic characteristics of Crouzon’s syndrome. (b) Lateral view of child demonstrating midface retrusion and exorbitism characteristic of Crouzon’s syndrome. (c) Superior view of infant illustrating site for bicoronal incisions. (d) Bilateral frontal bone plates are sectioned and removed.
(e) Barrel staving is accomplished to achieve frontocranial reshaping of the plates. (f) Supraorbital bar is removed and sectioned at
with an increased sebaceous discharge. Hydrocephalus occurs frequently and requires ventriculoperitoneal shunting.74–76 A conductive hearing loss may also be present.77,78
Timing and Surgical Management
The surgical management and timing are sequenced much the same as for patients with Crouzon’s syndrome (Figures 54.12c–h). The need for repeat cranio-orbital surgery to increase intracranial volume for the relief of intracranial pressure is greater. Mental retardation is more common and may
the midline. (g) Intraoperative superior view illustrating microplate fixation of the inner cortex and resulting increase in the intracranial volume. (h) Lateral intraoperative view demonstrating the cranial vault reshaping necessary prior to closure for correction of the brachycephalic deformity characteristic of Crouzon’s, Apert’s, or Pfeiffer’s syndromes.
be secondary to inadequate treatment of hydrocephalus or to craniosynostosis with reduced intracranial brain volume rather than inherent in the etiology itself.74–76
Pfeiffer Syndrome
Pfeiffer syndrome was first described in 1964. The clinical appearance of this syndrome is characterized by bilateral synostosis of the coronal sutures with associated midface deficiency, exorbitism, and exophthalmus (Figure 54.14). Broad thumbs, broad great toes, and partial soft tissue syndactyly of

54. Craniofacial Deformities: Introduction and Principles of Management |
681 |
e |
f |
g |
h |
FIGURE 54.12 Continued.
the hands are variable features. Hydrocephalus and intracra- |
branchial arch deformity, oculoauriculovertebral spectrum, |
nial hypertension have been reported in association with this |
otomandibular dysostosis, oculoauriculovertebral dysplasia, lat- |
syndrome. Intelligence is usually normal, but mental retarda- |
eral facial dysplasia, and unilateral craniofacial microsomia.80–85 |
tion has been described. |
It is the second most common congenital facial deformity after |
Timing and Surgical Management |
cleft lip and palate with an incidence of approximately 1 in 5600 |
live births.86 Although the etiology of HFM may be variable and |
|
The surgical management and timing are sequenced much the |
heterogenous, exposure of the pregnant mother to drugs such as |
same as for patients with Crouzon or Apert syndrome (Figure |
thalidomide, primidone, and retinoic acid has been associated |
54.12c–h). |
with a congenital first and second branchial arch syndrome.87–89 |
|
Hemifacial Microsomia (HFM)
The term hemifacial microsomia (HFM) was first used by Gorlin and Pindborg in 1964.79 Several other terms have also been used to describe this syndrome: asymmetric first and second
Pathogenesis
Typically, hemorrhage from the developing stapedial artery produces a hematoma in the area of the first and second branchial arches. The size of this hematoma and the resultant tissue destruction determines the morphology and variability

682 |
|
|
|
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a |
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
c
FIGURE 54.13 (a) Anterior view of a child with Apert’s syndrome. (b) Same child demonstrating characteristic syndactyly of the hand. (c) Same child demonstrating characteristic syndactyly of the foot.
of HFM as described in a experimental model by Poswillo.90 This sequence may be applicable to the condition in humans; additionally, hematoma formation may be the result of a variety of causes, such as hypoxia, hypertension, anticoagulants, or anomalous development of the carotid artery system.91
Clinical Characteristics of HFM
The mandible is short, retrusive, and narrow at birth and usually becomes progressively more asymmetric with time. The
mandibular malformation ranges from a small but normally shaped ramus and temporomandibular joint (TMJ) to complete absence of these structures. The midface normally grows downward and forward away from the cranial base. In HFM, the growth of maxilla on the affected side is decreased secondary to temporal bone abnormalities, mandibular hypoplasia, and neuromuscular defects. The affected maxilla is short and canting of the occlusal plane is present; the occlusal plane is tilted upward on the affected side.86,92,93
In untreated HFM, the abnormal mandible consists of a

54. Craniofacial Deformities: Introduction and Principles of Management |
683 |
|||||
a |
|
b |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.14 (a) Anterior view of an infant with Pfeiffer’s syndrome. (b) Lateral view of an infant with Pfeiffer’s syndrome.
short, medially displaced or absent ramus. If present, the ramus and mandibular body may be flat in contour and the chin is deviated toward the affected side. The occlusion on the affected side may be in crossbite and is generally tilted upward. The zygomatic bone is flat or incompletely formed, and the orbit may be inferiorly displaced.86,92,93
Soft Tissue Malformation
The soft tissue malformation consists of a decrease in the bulk of subcutaneous tissue ranging from mild to severe. The degree of soft tissue envelope deficit usually correlates with the severity of the skeletal defect. The soft tissue defects are classified as mild, moderate, and severe. The mild form consists of minimal subcutaneous and muscle hypoplasia, absence of or slight macrostomia, and a normal or mild auricular deformity. The severe form consists of significant subcutaneous and muscle hypoplasia, facial clefts, macrostomia, and neuromuscular weakness. Patients in between these two extremes are considered to have a moderate form. The external ear deformity is classified as grade I, II, and III following the system described by Meurman94:
Grade I. Mild hypoplasia, mild cupping, but all structures present.
Grade II. Absence of the external auditory canal and variable hypoplasia of the concha.
Grade III. Absence of the auricle, with an anteriorly and inferiorly displaced lobule.
A conductive hearing loss is present due to hypoplasia of the ear ossicles. Additionally, more than 25% of patients have cranial nerve abnormalities, usually consisting of facial nerve palsy and/or deviation of the palate toward the affected side with motion. Palatal deviation may be due to a combination of structural asymmetry, muscle hypoplasia, and cranial nerve weakness. The presence or absence of cranial nerve VII palsy correlates with the severity of the ear deficit, not the skeletal defect; the marginal mandibular nerve is the most common branch involved. Rarely, the total facial nerve palsy or a sensory deficit of trigeminal nerve has been described (Figure 54.15).95–97
Classification of HFM
Hemifacial microsomia has been classified into three types: type I, type II, and type III.92,93,96,97 This classification is based on the presence or absence of critical structures and also assists in treatment planning.
Type I
Skeletal
All components are present but hypoplastic to varying degrees. The TMJ is present, but the cartilage and joint space are reduced. Hinge movement is normal, but translation is reduced during jaw opening.

684 |
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a |
b |
FIGURE 54.15 (a) Anterior view of an infant with characteristics of moderate to severe hemifacial microsomia (HFM). (b) Lateral view of an infant with characteristics of HFM and a grade III ear deformity.
Muscle
All masticatory muscles are present but are small. Patterns of muscle use are within the normal range of variation.
Type II A
Skeletal
The movement of the TMJ is present but without translation. The morphology of the TMJ is abnormal. The condylar process is cone shaped and positioned anterior and medial to its normal position. The coronoid process and angle are well developed.
Muscle
All muscles are notably hypoplastic.
Type II B
Skeletal
No condylar process that articulates with the temporal bone is present, but the coronoid process of varying size is present.
Muscle
No lateral pterygoid muscle is attached to the TMJ. There are also deficiencies of the masseter and medial pterygoid muscle. The temporalis muscle is small but easily palpated and is attached to the coronoid process.
Type III
Skeletal
No condylar or coronoid process is present. Additionally, the angle is absent.
Muscular
Severe hypoplasia of the masticatory muscles is present. The lateral pterygoid and temporalis muscles are not attached to the mandible.
Treatment
In general, the treatment of HFM is divided into two groups: growing and nongrowing patients. The treatment for each group is discussed.
Treatment for the Growing Child
The overall objectives of treatment are:
1.Improved function
2.Optimal facial symmetry
3.Aesthetics
Treatment is directed toward four areas:
1.Increasing the size of the underdeveloped mandible and associated soft tissues
54. Craniofacial Deformities: Introduction and Principles of Management |
685 |
2.Building an articulation between the mandible and temporal bone
3.Correcting the maxillary deformity
4.Building an aesthetic appearance to the face and dentition
The sequence of treatment consists of the following:
1.Presurgical orthopedic jaw treatment
2.Lengthening of the mandibular ramus
3.Reconstruction of TMJ
4.Correction of the maxilla when necessary
5.Final orthodontic refinement and soft tissue augmentation
Presurgical Orthopedic Treatment
Type I
A functional appliance is constructed to hold the affected side of the mandible in a lowered, forward position. This is expected to stimulate an additional increase in the length of both the condylar and coronoid process. When treatment response is good, surgical lengthening may be avoided if the canting of the occlusal plane is acceptable.
Type II A
In this group, the canting of occlusal plane is severe. Lengthening of the mandible on the affected side is necessary and an open bite is created postoperatively. This open bite can be closed by active orthodontic extrusion of maxillary teeth. The need for a LeFort I osteotomy is usually avoided.
Type II B and Type III
In these groups, the mandibular condyle is missing; therefore, reconstruction of the TMJ with costochondral or sternoclavicular grafts is necessary. This is especially true when mandibular movement is restricted and masticatory function is impaired. Distraction osteogenesis may be considered for mandibular lengthening. This reconstruction should be done early in development (6 to 10 years of age). Additionally, a second mandibular lengthening may be necessary for correction of any residual deformities; often simultaneous maxillary surgery is also needed.
After reconstruction of the TMJ and lengthening of the mandible have been done in the growing child, a functional appliance is constructed to be used for continued growth management, thereby supporting the deficient joint structure and asymmetric muscle function.
Surgical Management
Type I and Type II A
Surgical correction of skeletal deformities in these groups is necessary in selected growing children with HFM. The mandibular lengthening with creation of an open bite is accomplished early in the mixed dentition stage. The mandible is elongated and rotated to the proper midline, leaving the
TMJ in place. Maxillary surgery is not necessary, and the created open bite on the affected side is maintained and regulated by an orthodontic appliance.92,96,97
Type II B and Type III
In these groups, the mandible is elongated and rotated by the construction of a mandibular ramus and TMJ with costochondral or sternoclavicular junction, iliac crest bone grafts, or both. The surgery and orthodontic procedures are otherwise the same as for patients with type I and type II A deformities.
Treatment for the Nongrowing Patient
Orthodontic Treatment
In the nongrowing patient with HFM, presurgical orthodontic treatment is necessary. The dentoalveolar adaptations to the asymmetry must be corrected, and coordination of arches is mandatory prior to surgery.
Surgical Management
Surgical treatment in the nongrowing patient with HFM consists of an operation to level the maxilla and piriform apertures, to make the mandible symmetric, and to place the TMJ in its proper location. In patients with type II B, the existing ramus is hypoplastic and located in such an abnormal position that it is not useful and must be excised and replaced. In type III HFM, a new TMJ and ramus of the mandible are constructed in the correct location (Figure 54.16). The auricular deformity may be reconstructed using autogenous or alloplastic materials.
Treacher Collins Syndrome (TCS)
Mandibulofacial Dysostosis
In 1889, Berry was the first to publish and describe this deformity in a 15-year-old girl who had notching in the outer region of the right lower lid.98 He commented on the possibility of hereditary transmission of this deformity. Treacher Collins recorded two cases in 1900 showing a more distinct development of this condition; the incidence is reported to be 1 in 10,000 live births.
Clinical Characteristics of TCS
This syndrome involves skeletal and soft tissue abnormalities of both the midface and lower face. It can be classified into three forms: complete, incomplete, and mild.99
Complete Form of TCS
In the orbital complex, there exists an absence or hypoplasia of the zygoma and overlying superficial musculoaponeurotic soft tissues. Malar prominence is absent and lateral canthal

a |
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
c |
d |
e
FIGURE 54.16
54. Craniofacial Deformities: Introduction and Principles of Management |
687 |
f |
g |
h |
|
|
|
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.16 Continued. (a) Anterior view of a nongrowing patient with characteristics of HFM. (b) Lateral view of a nongrowing patient with characteristics of HFM. (c) Mandible is elongated and rotated via placement of an autogenous costochondral graft. (d) Straightening genioplasty is accomplished for enhanced symmetry.
(e) Panoramic radiograph depicting the above procedures. Note
screws in left anterior maxilla stabilizing a silastic malar implant to help mask the soft tissue deformity. (f) Preauricular tags are excised and the ear deformity is reconstructed via osseointegrated dental implants (Stage II). (g) Suprastructure constructed and try-in done. (h) Anterior postoperative view. (i) Lateral postoperative view with auricular prosthesis in place.

688 |
|
|
|
|
|
G.E. Ghali, W. Tharanon, and D.P. Sinn |
a |
|
|
|
|
|
b |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
c
FIGURE 54.17 (a) Anterior view of child with Treacher Collins syndrome. (b) Lateral view of child with TCS. (c) Autogenous cranial bone grafts used to reconstruct orbits and zygomas. (d) Placement of graft into zygomatico-orbital region. (e) Anterior long-term post-
dystopia, upper eyelid pseudoptosis, and antimongoloid slant of the palpebral fissure may be observed. There is usually a coloboma or marginal hypoplasia of the lower eyelid.
In the maxillomandibular complex, abnormalities of both jaws are noted. Shortness of the maxilla with narrowing of the palate and choanal atresia are often present. Shortness of the ramus and body of the mandible results in retrognathia.
Middle ear abnormalities and microtia may cause hearing loss with subsequent impairment of speech and intellectual development.
Incomplete Form of TCS
All the deformities are present but to a milder form.
operative view following correction of TCS with autogenous cranial bone grafts (CBG). (f) Lateral long-term postoperative view following correction of TCS with autogenous CBG.
Mild Form of TCS
Generally, the orbitotemporal region is more affected than are the maxilla or mandible.
Timing and Surgical Treatment
Principles of treatment are as follows:
1.Hearing aids should be used as early as possible.
2.Correction of eyelid colobomas should be performed in the first few years of life. These are repaired early because at a later stage the orbits are dissected and bone grafts placed producing tension on the soft tissues that may not allow their optimal closure.

54. Craniofacial Deformities: Introduction and Principles of Management |
689 |
d
e |
|
|
|
|
|
f |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 54.17 Continued.
3.Auricular deformities should be corrected after 8 years of age because substantial autogenous rib cartilage is available.
4.Orthognathic surgery, if needed, should be performed between 6 and 10 years of age and only when the child has major breathing problems.
5.Orbital surgery should be performed within the same age
range (6 to 10 years). We prefer cranial bone to reconstruct the orbits and zygomatic arches because, in our experience, resorption is less than with rib or iliac crest (Figure 54.17). Vascularized cranial bone grafts may be used as an alternative to reconstruct the malar bone. Preliminary results have shown that the resorption is less than with free cranial bone grafts.
690
Summary
In approximately 1 in 1000 births in the United States the infant has a variant of some facial, skeletal, or craniofacial deformity. If cleft lip and palate deformities are included, the incidence is far greater.
The surgical approach to a majority of these congenital deformities was radically changed by techniques introduced by Paul Tessier in France in 1967. From his imaginative intracranial and extracranial approaches, numerous advances have been made that facilitate the care of the majority of these children. More recently additional advances in pediatric anesthesia, bioresorbable plating systems, and distraction osteogenesis have improved the management of these patients.100–103
Timing of the surgical management of these patients has been advocated from the first few weeks after birth until well into the second decade. Many of these patients will need multiple, staged procedures involving movements of bone and soft tissue from both an intracranial and extracranial approach.
Acknowledgments: The authors would like to express their appreciation to the members of the craniofacial team at Dallas Children’s Medical Center for contributing to the treatment of these patients (H.S. Byrd, MD, Craig Hobar, MD, D.P. Sinn, DDSc, and Fred Sklar, MD).
References
1.Johnston MC. Embryology of the head and neck. In: McCarthy JG, ed. Plastic Surgery. Philadelphia: WB Saunders; 1990: 2451–2495.
2.Sperber GH. Craniofacial Embryology, 4th Ed. London: Wright; 1989.
3.Sadler TW. Langman’s Medical Embryology, 6th Ed. Baltimore: Williams & Wilkins; 1990.
4.Slavkin HC. Developmental Craniofacial Biology. Philadelphia: Lea & Febiger; 1979.
5.Sulik KK, Cook CS, Webster WS. Teratogens and craniofacial malformations: relationships to cell death. Development. 1988; 103:213–232.
6.Marsh JL, Vannier MW. Comprehensive Care for Craniofacial Deformities. St. Louis: CV Mosby; 1985.
7.Brodsky L, Ritter-Schmidt DH, Holt L. Craniofacial Anomalies: An Interdisciplinary Approach. St. Louis: Mosby Yearbook; 1989.
8.Gordon R, Herman GT, Johnston SA. Image reconstruction from projections. Sci Am. 1975;233:56.
9.Robb RA. X-ray computer tomography: an engineering synthesis of multiscientific principles. In: Bourne JR, ed. Critical Reviews in Biomedical Engineering. Boca Raton, FL: CRC Press; 1982.
10.Ter-Pogessian MN. Computerized cranial tomography: equipment and physics. Semin Roentgenol. 1977;12:13.
11.Marsh JL, Gado M. Surgical anatomy of the craniofacial dysostosis: insights from CT scans. Cleft Palate J. 1982;19:212.
12.Cutting C. Computer-aided planning and evaluation of facial and orthognathic surgery. Clin Plast Surg. 13:449.
G.E. Ghali, W. Tharanon, and D.P. Sinn
13.Cutting C, Bookstein FL, Grayson B, Fellingham L, McCarthy JG. Three-dimensional computer-assisted design of craniofacial surgical procedures: optimization and interaction with cephalometric and CT-based models. Plast Reconstr Surg. 1986;77:877–887.
14.Marsh JL, Gado M. The longitudinal orbital CT projection: a versatile image for orbital assessment. Plast Reconst Surg. 1983;71:308.
15.Marsh JL, Vannier MW. The “third” dimension in craniofacial surgery. Plast Reconstr Surg. 1983;71:759.
16.Vannier MW, Marsh JL, Warren JO. Three dimension CT reconstruction images for craniofacial surgical planning and evaluation. Radiology. 1984;150:179.
17.Marsh JL. Computerized imaging for soft tissue and osseous reconstruction in the head and neck. Clin Plast Surg. 1985;12:279.
18.Marsh JL. Applications of computer graphics in craniofacial surgery. Clin Plast Surg. 1986;13:441.
19.Marsh JL, Vanier MW. Computer-assisted imaging in the diagnosis, management and study of dysmorphic patients. In: Vig KWL, Burdi AR, eds. Craniofacial Morphogenesis and Dysmorphogenesis. Ann Arbor, MI: University of Michigan Press; 1988:109–126.
20.Marsh JL, Vannier MW. Three-dimensional surface imaging from CT scans for the study of craniofacial dysmorphology.
J Craniofac Genet Dev Biol. 1989;9:61.
21.Lo LJ. Craniofacial computer-assisted surgical planning and simulation. Clin Plast Surg. 1994;21:501.
22.Altobelli DE. Computer-assisted three-dimension planning in craniofacial surgery. Plast Reconstr Surg. 1993;92:576.
23.Posnick JC. Indirect intracranial volume measurements using CT scans: clinical applications for craniosynostosis. Plast Reconstr Surg. 1992;89:34.
24.Posnick JC, Goldstein JA, Waitzman AA. Surgical correction of the Treacher Collins malar deficiency: quantitative CT scan analysis of long-term results. Plast Reconstr Surg. 1992;92:12.
25.Oakes WJ. Craniosynostosis. In: Serafin D, Geargiade NG, eds. Pediatric Plastic Surgery. St. Louis: CV Mosby; 1984:404– 439.
26.Virchow R. Uberden Cretinismus, namentlich in Franken, und uber pathologische schädelformen. Verh Phys Med Gesellsch Uurzb. 1851;2:230.
27.Persing JA, Jane JA, Shaffrey M. Virchow and the pathogenesis of craniosynostosis: a translation of his original work. Plast Reconstr Surg. 1989;83:738.
28.Moss ML. The pathogenesis of premature cranial synostosis in man. Acta Anat (Basel). 1959;37:351.
29.Albright AL, Byrd RP. Suture pathology in craniosynostosis. J Neurosurg. 1981;54:384.
30.Graham JM, de Saxe M, Smith DW. Sagittal craniosynostosis: fetalheadconstraintasonepossiblecause.J Pediatr. 1979;95:747.
31.Renier D, Sainte-Rose C, Marchac D, et al. Intracranial pressure in craniostenosis. J Neurosurg. 1982;57:370.
32.Goldstein SJ, Kidd RC. Value of computed tomography in the evaluation of craniosynostosis. Comput Radiol. 1982;6:331–336.
33.Gault D, Brunelle F, Renier D, et al. The calculation of intracranial volume using CT scans. Child’s Nerv Syst. 1988;4:271.
34.Posnick JC, Bite U, Nakano P, et al. Comparison of direct and indirect intracranial volume measurements. Proceedings of the 6th International Congress on Cleft Palate and Related Craniofacial Anomalies; June 1989.
54. Craniofacial Deformities: Introduction and Principles of Management |
691 |
35.Patton MA, Goodship J, Hayward R, et al. Intellectual development in Apert’s syndrome: a long term follow-up of 29 patients. J Med Genet. 1988;25:164.
36.Arndt EM. Psychosocial adjustment of 20 patients with Treacher Collins syndrome before and after reconstructive surgery. Br J Plast Surg. 1987;40:605.
37.Ousterhout DK, Vargervik K. Aesthetic improvement resulting from craniofacial surgery in craniosynostosis syndromes.
J Craniomaxillofac Surg. 1987;15:189.
38.Barden RC. The physical attractiveness of facially deformed patients before and after craniofacial surgery. Plast Reconstr Surg. 1988;82:229.
39.Barden RC. Emotional and behavioral reactions to facially deformed patients before and after craniofacial surgery. Plast Reconstr Surg. 1988;82:409.
40.Edgerton MT, Jane JA, Berry FA, et al. The feasibility of craniofacial osteotomies in infants and young children. Scand J Plast Reconstr Surg. 1974;8:164.
41.Edgerton MT, Jane JA, Berry FA. Craniofacial osteotomies and reconstruction in infants and young children. Plast Reconstr Surg. 1974;54:13.
42.Hoffman HJ, Mohr G. Lateral canthal advancement of the supraorbital margin: a new corrective technique in the treatment of coronal synostosis. J Neurosurg. 1976;45:376.
43.Jane JA, Park TS, Zide BM, et al. Alternative techniques in the treatment of unilateral coronal synostosis. J Neurosurg. 1984;61:550.
44.Marchac D. Forehead remolding for craniosynostosis. In: Converse JM, McCarthy JG, Wood-Smith D, eds. Symposium on Diagnosis and Treatment of Craniofacial Anomalies. St. Louis: CV Mosby; 1979:323.
45.Marchac D, Renier D. Treatment of craniosynostosis in infancy. Clin Plast Surg. 1987;14:61.
46.Marchac D, Renier D, Jones BM. Experience with the “floating forehead.” Br J Plast Surg. 1988;41:1.
47.McCarthy JG. New concepts in the surgical treatment of the craniofacial synostosis syndromes in the infant. Clin Plast Surg. 1979;6:201.
48.McCarthy JG, Coocaro PJ, Epstein F, et al. Early skeletal release in the infant with craniofacial dysostosis: the role of the sphenozygomatic suture. Plast Reconst Surg. 1978;62:335.
49.McCarthy JG, Epstein F, Sadove M, et al. Early surgery for craniofacial synostosis: an 8-year experience. Plast Reconstr Surg. 1984, 73:532.
50.Persing JA, Babler WJ, Jane JA, et al. Experimental unilateral coronal synostosis in rabbits. Plast Reconst Surg. 1986;78:594.
51.Tressera L, Fuenmayor P. Early treatment of craniofacial deformities. J Maxillofac Surg. 1981;9:7.
52.Whitaker LA, Schut L, Kerr LP. Early surgery for isolated craniofacial dysostosis. Plast Reconstr Surg. 1977;60:575.
53.Whitaker LA, Barlett SP, Schut L, et al. Craniosynostosis: an analysis of the timing, treatment and complication in 164 patients. Plast Reconstr Surg. 1987;80:195.
54.Barden RC, Ford ME, Jensen AG, et al. Effects of craniofacial deformity in infancy on the quality of mother-infant interaction. Child Dev. 1989;60:819.
55.Lefebvre A, Barclay S. Psychosocial impact of craniofacial deformities before and after reconstructive surgery. Can J Psychiatry. 1982;27:579.
56.Lefebvre A, Travis F, Arndt EM, et al. A psychiatric profile
before and after reconstructive surgery in children with Apert’s syndrome. Br J Plast Surg. 1986;39:510.
57.Palkes HS, Marsh JL, Talent BK. Pediatric craniofacial surgery and parental attitudes. Cleft Plate J. 1986;23:137.
58.Muhling J, Reuther J, Sorensen N. Problems with lateral canthal advancement. Child’s Nerv Syst. 1986;2:287.
59.Longacre JJ, Destafano GA, Holmstrand K. The early versus the late reconstruction of congenital hypoplasia of the facial skeleton and skull. Plast Reconstr Surg. 1961;27:489.
60.McCarthy JG, Coccaro PJ, Epstein FJ. Early skeletal release in the patient with craniofacial dysostosis. In: Converse JM, McCarthy J, Wood-Smith D, eds. Symposium on Diagnosis and Treatment of Craniofacial Anomalies. St. Louis: CV Mosby; 1979:295.
61.McLaurin RL, Matson DD. Importance of early surgical treatment of craniosynostosis. Pediatrics. 1952;10:637.
62.Mohr G, Hoffman HJ, Munro IR, et al. Surgical management of unilateral and bilateral coronal craniosynostosis: 21 years of experience. Neurosurgery. 1978;2:83.
63.Shillito J, Matson DD. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41:829.
64.Tessier P. The definitive plastic surgical treatment of the severe facial deformities of craniofacial dysostosis. Crouzon’s and Apert’s disease. Plast Reconstr Surg. 1971;48:419.
65.Kreiborg S, Bjork A. Description of a dry skull with Crouzon syndrome. Scand J Plast Reconstr Surg. 1982;16:245–253.
66.Carinci F, Avantaggiato A, Curioni A. Crouzon syndrome: cephalometric analysis and evaluation of pathogenesis cleft palate. Craniofacial J. 1994;31:201–209.
67.Schiller JG. Craniofacial dysostosis of Crouzon: a case report and pedigree with emphasis on heredity. Pediatrics. 1959;23: 107.
68.Nakano PH, Posnick JC. Long-term results of reconstructive craniofacial surgery in patients with Crouzon and Apert syndrome. Proceedings of the 46th annual meeting of the Cleft Palate Craniofacial Association; April 1989.
69.Nakano PH, Posnick JC. Long-term results of reconstruction in craniofacial dysostosis. Proceedings of the 6th International Congress of Cleft Palate and Related Anomalies; June 1989.
70.Wheaton SW. Two specimens of congenital cranial deformities in infants in association with fusion of the fingers and toes. In: Smith DW, ed. Recognizable Patterns of Human Malformation, Major Problems in Clinical Pediatrics, Vol VII. Philadelphia: WB Saunders; 1982:308.
71.Tessier P. Apert’s syndrome: acrocephalosyndactyly Type I. In: Caronni EP, ed. Craniofacial Surgery. Boston: Little, Brown, 1985;280–303.
72.Tessier P. Relationship of craniostenosis to craniofacial dysostosis and to faciostenosis. A study with therapeutic implications. Plast Reconstr Surg. 1971;48:224.
73.Pollard ZF. Bilateral superior oblique muscle palsy associated with Apert’s syndrome. Am J Ophthalmol. 1988;106:337.
74.Fishman MA, Hogan GR, Dodge PR. The concurrence of hydrocephalus and craniosynostosis. J Neurosurg. 1971;34:621.
75.Golabi M, Edwards MSB, Ousterhout DK. Craniosynostosis and hydrocephalus. Neurosurgery. 1987;21:63.
76.Hogan GR, Bauman ML. Hydrocephalus in Apert’s syndrome. J Pediatr. 1971;79:782.
77.Alberti PW, Ruben RJ. Otologic Medicine and Surgery. New York: Churchill Livingstone; 1988.
692
78.Corey JP, Caldarelli DD, Gould HJ. Otopathology in cranial facial dysostosis. Am J Otol. 1987;8:14.
79.Gorlin RJ, Pindborg J. Syndromes of the Head and Neck. New York: McGraw-Hill; 1964:261–265; 419–425.
80.Cohen MM, Rollnick BR, Kaye CI. Oculoauriculovertebral spectrum: an update critique. Cleft Palate J. 1989;26:276.
81.Francois J, Haustrate L. Anomalies colobomateuses du globe oculair et syndrome du premier arc. Ann Ocul. 1954;187:340.
82.Gorlin RJ, Jue KL, Jacobson NP, et al. Oculoauriculovertebral dysplasia. J Pediatr. 1963;63:991.
83.Grabb WC. The first and second branchial arch syndrome.
Plast Reconstr Surg. 1965;36:485.
84.Grayson BH, Boral S, Eisig S, et al. Unilateral craniofacial microsomia. I. Mandibular analysis. Am J Orthod. 1983;84:225.
85.Ross RB. Lateral facial dysplasia (first and second branchial arch syndrome, hemifacial microsomia). Birth Defects. 1975;11:51.
86.Kaban LB, Mulliken JB, Murray JE. Three dimensional approach to analysis and treatment of hemifacial microsomia.
Cleft Palate J. 1981;18:90.
87.Gustavson EE, Chen H. Goldenhar syndrome, anterior encephalocele and aqueductal stenosis following fetal primidone exposure. Teratology. 1985;32:13.
88.Lammer EJ, Chen DT, Hoar RM, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313:837.
89.Miehlke A, Partsch CJ. Ohrmissbilding, fascialis und abducenslahmung als syndrome der thalidomidschadigung. Arch Ohrenheilkd. 1963;181:154.
90.Poswillo D. The pathogenesis of first and second branchial arch syndrome. Oral Surg. 1973;35:302.
91.Soltan HC, Holmes LB. Familial occurrence of malformations possibly attributable to vascular abnormalities. J Pediatr. 1986; 109:112.
G.E. Ghali, W. Tharanon, and D.P. Sinn
92.Murray JE, Kaban LB, Mulliken JB. Analysis and treatment of hemifacial microsomia. Plast Reconstr Surg. 1984;74: 186.
93.Murray JE, Kaban LB, Mulliken JB, et al. Analysis and treatment of hemifacial microsomia. In: Caronni EP, ed. Craniofacial Surgery. Boston: Little, Brown; 1985:377–390.
94.Meurman Y. Congenital microtia and meatal atresia. Arch Otolaryngol. 1957;66:443.
95.Bennun RD, Mulliken JB, Kaban LB, et al. Microtia: a microform of hemifacial microsomia. Plast Reconstr Surg. 1985;76:859.
96.Kaban LB, Moses ML, Mulliken JB. Correction of hemifacial microsomia in the growing child. Cleft Palate J. 1986;23:50.
97.Kaban LB, Moses ML, Mulliken JB. Surgical correction of hemifacial microsomia in the growing child. Plast Reconstr Surg. 1988;82:9.
98.Berry GA. Note on a congenital defect (? coloboma) of the lower lid. Ophthalmol Hosp Rep. 1889;12:255.
99.Tessier P. Tulasne JF. Surgical correction of Treacher Collins Syndrome. In: Bell WH, ed. Modern Practice in Orthognathic and Reconstructive Surgery. Philadelphia: WB Saunders; 1992:1601–1623.
100.Kurpad SN, Goldstein JA, Cohen AR. Bioresorbable fixation for congenital pediatric craniofacial surgery: a 2 year followup. Pediatr Neurosurg. 2000;33:306–310.
101.McCarthy JG, Stelnicki EJ, Grayson BH. Distraction osteogenesis of the mandible: a ten year experience. Semin Orthod. 1999;5:3–8.
102.Tate GS, Tharanon W, Sinn DP. Transoral maxillary distrac-
tion osteogenesis of an unrepaired |
bilateral alveolar cleft. |
J Craniofac Surg. 1999;10:369–374. |
|
103. Cohen SR. Midface distraction. Semin Orthod. 1999;5:52–58.