

Wasserscheid P., Welton T. - Ionic Liquids in Synthesis (2002)(en)
.pdf206 Martyn Earle
O
acetyl chloride
H3C
[EMIM]Cl-AlCl3 (X = 0.67) 5 min, 0°C
Scheme 5.1-63: The acetylation of 1,1,2,6-tetramethyl-3-isopropylindane in [EMIM]Cl/AlCl3 (X(AlCl3) = 0.67).
H3CCOCl |
O |
[EMIM]Cl-(AlCl3) (X = 0.67) |
|
fast
[H3CCO]+ slow |
+ |
O |
O |
[H3CCO]+
O  O O
O O
+
major isomer |
minor isomer |
O
Scheme 5.1-64: The acetylation of anthracene in [EMIM]Cl/AlCl3 (X(AlCl3) = 0.67).
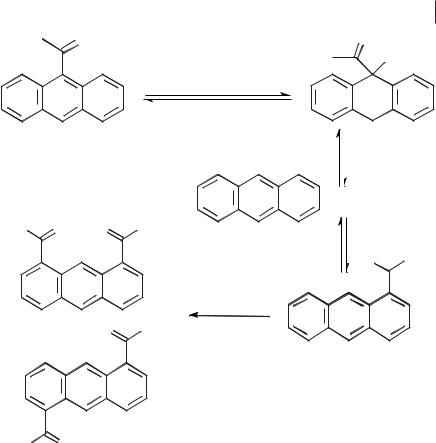
This was confirmed by taking a sample of 9-acetylanthracene and allowing it to isomerize in the ionic liquid. This gave a mixture of anthracene, 1,5-diacetylan- thracene and 1,8-diacetylanthracene. It should be noted that a proton source was needed for this reaction to occur, implying an acid-catalyzed mechanism (Scheme 5.1-65) [95].
5.1 Stoichiometric Organic Reactions and Acid-Catalyzed Reactions in Ionic Liquids 207
O |
O |
H+ / |
H |
[EMIM]Cl-AlCl3 X = 0.67)
|
|
|
|
|
|
|
+ |
||
|
|
|
|
|
|
|
|
|
|
+ |
H C |
|
C |
|
O |
|
|||
|
|
|
|||||||
|
3 |
|
|
|
|
|
|
||
H3C |
O O |
CH3 |
O CH3
CH3
[H3CCO]+ |
+ H+ |
H+ + |
+ O |
CH |
|
|
3 |
H3C O
Scheme 5.1-65: Proposed mechanism for the isomerization of 9-acetylanthracene in [EMIM]Cl/AlCl3 (X(AlCl3) = 0.67).
The Friedel–Crafts acylation reaction has also been performed in iron(III) chloride ionic liquids, by Seddon and co-workers [96]. An example is the acetylation of benzene (Scheme 5.1-66). Ionic liquids of the type [EMIM]Cl/FeCl3 (0.50 < X(FeCl3) < 0.62) are good acylation catalysts, with the added benefit that the ketone product of the reaction can be separated from the ionic liquid by solvent extraction, provided that X(FeCl3) is in the range 0.51–0.55.
The ability of iron(III) chloride genuinely to catalyze Friedel–Crafts acylation reactions has also been recognized by Hölderich and co-workers [97]. By immobilizing the ionic liquid [BMIM]Cl/FeCl3 on a solid support, Hölderich was able to acetylate mesitylene, anisole, and m-xylene with acetyl chloride in excellent yield. The performance of the iron-based ionic liquid was then compared with that of the corresponding chlorostannate(II) and chloroaluminate(III) ionic liquids. The results are given in Scheme 5.1-67 and Table 5.1-5. As can be seen, the iron catalyst gave superior results to the aluminiumor tin-based catalysts. The reactions were also carried out in the gas phase at between 200 and 300 °C. The acetylation reac-
208 |
Martyn Earle |
|
|
|
|
|
|
|
|
|
||||
|
|
|
O |
|
|
|
|
|
|
O |
||||
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
[EMIM]Cl-FeCl3 |
(X = 0.53) |
|
|
|
|
|
CH3 + HCl |
|||
|
|
+ |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
Cl |
CH3 |
|
|
|
|
|
|
|
|||
|
|
|
Scheme 5.1-66: |
The acetylation of benzene in an iron(IIII) chloride-based ionic liquid. |
||||||||||
tion was complicated by two side reactions. In the reaction between acetyl chloride and m-xylene, for example, the decomposition of acetyl chloride to ketene and the formation of 1-(1-chlorovinyl)-2,4-dimethylbenzene were also found to occur [97].
Rebeiro and Khadilkar have investigated the reactions between trichloroalkanes and aromatic compounds. For example, the benzoylation of aromatic compounds in ionic liquids was performed with benzotrichloride, giving ketones on aqueous workup [98].
5.1.2.4Cracking and isomerization reactions
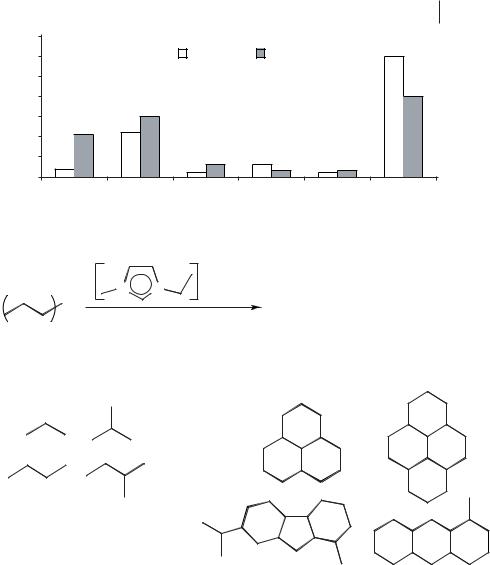
Cracking and isomerization reactions occur readily in acidic chloroaluminate(III) ionic liquids. A remarkable example of this is the reaction of poly(ethene), which is converted into a mixture of gaseous alkanes of formula (CnH2n+2, where n = 3–5) and cyclic alkanes with a hydrogen to carbon ratio of less than two (Figure 5.1-4, Scheme 5.1-68) [99].
|
O |
|
|
|
|
|
O |
|
|||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
||||
+ |
|
|
|
[BMIM]Cl-FeCl3 |
(X = 0.67) |
|
|
|
|
|
CH3 |
+ HCl |
|
|
|
CH3 FK 700 amorphous silica |
|
|
|
|
|
||||||
Cl |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Scheme 5.1-67: The acetylation of aromatics with supported ionic liquids (FK 700 is a type of amorphous silica made by Degussa).
Table 5.1-5: The acylation of aromatics in batch reactions at 100 °C, for 1 hour. Ratio of aromatic compound to acetylating agent = 5:1, mes. = mesitylene.
Ionic liquid |
Reaction |
Molar ratio |
% |
% |
|
|
IL : Ar-H |
Conversion |
Selectivity |
|
|
|
|
|
[BMIM]Cl/AlCl3 |
mes. + AcCl |
1:205 |
68.1 |
98 |
[BMIM]Cl/AlCl3 |
anisole + Ac2O |
1:45 |
8.3 |
96 |
[BMIM]Cl/AlCl3 |
m-xylene + AcCl |
1:205 |
3.5 |
96 |
[BMIM]Cl/FeCl3 |
mes. + AcCl |
1:205 |
94.7 |
95 |
[BMIM]Cl/FeCl3 |
anisole + Ac2O |
1:45 |
100 |
98 |
[BMIM]Cl/FeCl3 |
m-xylene + AcCl |
1:205 |
33.8 |
79 |
[BMIM]Cl/SnCl2 |
anisole + Ac2O |
1:45 |
19.7 |
94 |
[BMIM]Cl/SnCl2 |
m-xylene + AcCl |
1:205 |
3.6 |
95 |
5.1 Stoichiometric Organic Reactions and Acid-Catalyzed Reactions in Ionic Liquids 209
|
70 |
|
120 °C |
200 °C |
|
|
60 |
|
|||
|
50 |
|
|
|
|
yield |
40 |
|
|
|
|
30 |
|
|
|
|
|
|
|
|
|
|
|
|
20 |
|
|
|
|
|
10 |
|
|
|
|
|
0 |
|
|
|
|
|
propane |
isobutane |
butane |
isopentane |
other-volatile low-volatile |
Figure 5.1-4: The products from the ionic liquid cracking of high-density polyethylene at 120 °C and at 200 °C.
N |
+ N |
[Al2Cl7]- |
n |
Proton Source |
|
Examples of
Volatile Products
Volatile products CaH(2a+2)
+
Low-volatile products CbH(<2b)
Examples of Low-
Volatile Products
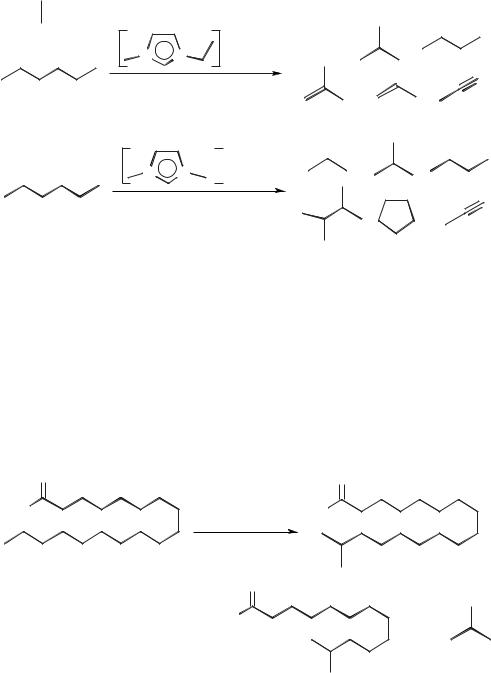
Scheme 5.1-68: The cracking of poly(ethene) in an ionic liquid.
The distribution of the products obtained from this reaction depends upon the reaction temperature (Figure 5.1-4) and differs from those of other poly(ethene) recycling reactions in that aromatics and alkenes are not formed in significant concentrations. Another significant difference is that this ionic liquid reaction occurs at temperatures as low as 90 °C, whereas conventional catalytic reactions require much higher temperatures, typically 300–1000 °C [100]. A patent filed for the Secretary of State for Defence (UK) has reported a similar cracking reaction for lower molecular weight hydrocarbons in chloroaluminate(III) ionic liquids [101]. An
210 Martyn Earle
N + N |
- |
CH4 |
[Al2Cl7] |
|
N + N 
 [Al2Cl7]-
[Al2Cl7]-
CuCl2
Scheme 5.1-69: The cracking of hexane in [EMIM]Cl/AlCl3 (X(AlCl3) = 0.67) with and without added copper(II) chloride.
example is the cracking of hexane to products such as propene and isobutene (Scheme 5.1-69). The reaction was also performed with added copper(II) chloride, which gave a significantly different product distribution.
A similar reaction occurs with fatty acids (such as stearic acid) or methyl stearate, which undergo isomerization, cracking, dimerization, and oligomerization reactions. This has been used to convert solid stearic acid into the more valuable liquid isostearic acid [102] (Scheme 5.1-70). The isomerization and dimerization of oleic acid and methyl oleate have also been found to occur in chloroaluminate(III) ionic liquids [103].
O |
|
O |
RO |
[EMIM][Al2Cl7] |
RO |
|
|
R = H, CH3
O
RO
+
+
Scheme 5.1-70: Cracking and isomerization of fatty acids and fatty acid methyl esters in chloroaluminate(III) ionic liquids.

5.1 Stoichiometric Organic Reactions and Acid-Catalyzed Reactions in Ionic Liquids 211
References
1 |
T. Welton, Chem. Rev. 1999, 99, |
20 |
E. Nakamura, M. Shimizu, I. Kuwaji- |
|
2071–2083. |
|
ma, J. Sakata, K. Yokoyama, R. Noyori, |
2 |
J. D. Holbrey. K. R. Seddon, Clean |
|
J. Org. Chem. 1983, 48, 932–945. |
|
Prod. Proc. 1999, 1, 223–236. |
21 |
G. Majetich, A. Casares, D. Chapman, |
3 |
M. J. Earle, K. R. Seddon, Pure Appl. |
|
M. Behnke, J. Org. Chem. 1986, 51, |
|
Chem. 2000, 72, 1391–1398. |
|
1745–1753. |
4 |
R. M. Pagni, in Advances in Molten |
22 |
T. W. Green, P. G. M. Wuts, Protective |
|
Salt Chemistry, Vol. 6 (G. Mamantov, |
|
Groups in Organic Synthesis, Wiley, |
|
J. Braunstein eds.), Elsevier, Oxford, |
|
New York, 1991. |
|
1987, 211–346. |
23 |
D. P. Cox, J. Terpinski, W. Lawry- |
5 |
D. W. Rooney, K. R. Seddon, in The |
|
nowicz, J. Org. Chem. 1984, 49, |
|
Handbook of Solvents (G. Wypych ed.), |
|
3216–3219. |
|
ChemTech Publishing, New York, |
24 |
R. A. Moss, W. Lawrynowicz, J. Org. |
|
2001, 1459–1484. |
|
Chem. 1984, 49, 3828–3830. |
6 |
K. R. Seddon, J. Chem. Tech. Biotech. |
25 |
D. P. Cox, R. A. Moss, J. Terpinski, J. |
|
1997, 68, 351–356. |
|
Am. Chem. Soc. 1983, 105, 6513–6514. |
7 |
K. R. Seddon, Green Chem, 2002, 4, |
26 |
P. Studt, Lebigs Ann. Chem. 1978, |
|
147–151. |
|
528–529. |
8 |
P. Wasserscheid, W. Keim, Angew. |
27 |
K. Nakasuji, K. Yoshida and I. Murata, |
|
Chem. Int. Ed. 2000, 39, 3773–3789. |
|
J. Am. Chem. Soc. 1983, 105, |
9 |
J. Dupont, C. S. Consorti, J. Spencer, |
|
5136–5137. |
|
J. Braz. Chem. Soc. 2000, 11, 337–344. |
28 |
M. B. Groen, H. Schadenberg, H. |
10 |
R. A. Sheldon, Chem. Commun. 2001, |
|
Wynberg, J. Org. Chem. 1971, 36, |
|
2399–2407. |
|
2797–2809. |
11 |
M. Fremantle, Chem. Eng. News 1998 |
29 |
H. Imaizumi, S. Sekiguchi, K. Matsui, |
|
(30th March), 76, 32–37. |
|
Bull. Chem. Soc. Jpn. 1977, 50, |
12 |
C. M. Gordon, J. D. Holbrey, A. R. |
|
948–952. |
|
Kennedy, K. R. Seddon, J. Mater. |
30 |
A. C. Buchanan III, D. M. Chapman, |
|
Chem. 1998, 8, 2627–2636. |
|
G. P. Smith, J. Org. Chem. 1985, 50, |
13 |
K. R. Seddon, A. Stark, M. J. Torres, |
|
1702–1711. |
|
Pure App. Chem. 2000, 72, 2275–2287. |
31 |
G. P. Smith, A. S. Dworkin, R. M. |
14 |
A. E. Visser, R. P. Swatloski, R. D. |
|
Pagni, S. P. Zingg, J. Am. Chem. Soc. |
|
Rogers, Green Chem. 2000, 2, 1–4. |
|
1989, 111, 525–530. |
15 |
J. March, Advanced Organic Chemistry, |
32 |
C. J. Adams, M. J. Earle, K. R. Seddon, |
|
4th ed., Wiley, Chichester, 1992. |
|
Chem. Commun. 1999, 1043–1044. |
16 |
M. Fieser, L. Fieser, Reagents for |
33 |
L. Green, I. Hemeon, R. D. Singer, |
|
Organic Synthesis, Vol. 1, Wiley-Inter- |
|
Tetrahedron Lett. 2000, 41, 1343–1345. |
|
science, New York, 1967, 936, and ref- |
34 |
Z. Ma, Y. Q. Deng, F. Shi, Can. Patent |
|
erences therein. |
|
CN 1247856, 2000. |
17 |
C. M. Suter, The Organic Chemistry of |
35 |
Y. Q. Deng, F. Shi, J. J. Beng, K. Qiao, |
|
Sulfur, Tetravalent Sulfur Compounds, |
|
J. Mol. Cat. A 2001, 165, 33–36. |
|
Wiley-Interscience, New York, 1944, |
36 |
C. W. Lee, Tetrahedron Lett. 1999, 40, |
|
420. |
|
2461–2462. |
18 |
G. P. Smith, R. M. Pagni in Molten |
37 |
J. D. Huddleston, H. D. Willauer, |
|
Salt Chemistry, An introduction to select- |
|
R. P. Swatloski, A. E. Visser, R. D. |
|
ed Applications (G. Mamantov, R. |
|
Rogers, Chem. Commun. 1998, |
|
Marassi eds.), D. Reidel Publishing |
|
1765–1766. |
|
Co., Dordrecht, 1987, 383–416. |
38 |
A. J. Carmichael, M. J. Earle, J. D. |
19 |
R. K. Sharma, J. L. Fry, J. Org. Chem. |
|
Holbrey, P. B. McCormac, K. R. Sed- |
|
1983, 48, 2112–2114. |
|
don, Org. Lett. 1999, 1, 997–1000. |
212 |
Martyn Earle |
|
|
|
|
|
|
L. P. Barthel–Rosa, J. A. Gladysz, |
62 |
J. Peng, Y. Deng, Tetrahedron Lett. |
|
39 |
|||||
|
|
Coord. Chem. Rev. 1999, 192, 587–605. |
|
2001, 42, 5917–5919. |
|
40 |
D. A. Jaeger, C. E. Trucker, Tetra- |
63 |
R. D. Singer, P. J. Scammells, Tetra- |
||
|
|
hedron Lett. 1989, 30, 1785–1788. |
|
hedron Lett. 2001, 42, 6831–6833. |
|
41 |
J. Howarth, K. Hanlon, D. Fayne, P. B. |
64 |
S. V. Ley, A. G. Leach, R. I. Storer, |
||
|
|
McCormac, Tetrahedron. Lett. 1997, |
|
J. Chem. Soc., Perkin Trans. 1 2001, |
|
|
|
38, 3097–3099. |
|
|
358–361. |
42 |
M. J. Earle, P. B. McCormac, K. R. |
65 |
C. Hardacre, J. D. Holbrey, S. E. |
||
|
|
Seddon, Green Chem. 1999, 1, 23–25. |
|
McMath, Chem. Commun. 2001, |
|
43 |
M. J. Earle, in Ioniy Liquids, R. D. |
|
367–368. |
||
|
|
Rogers, K. R. Seddon eds., ACS Sym- |
66 |
J. A. Boon, S. W. Lander Jr., J. A. |
|
|
|
posium Series, 2002, 818, 90–105. |
|
Levisky, J. L. Pflug, L. M. Skrznecki- |
|
44 |
T. Fisher, A. Sethi, T. Welton, J. Woolf, |
|
Cooke, J. S. Wilkes, Proceedings of the |
||
|
|
Tetrahedron Lett. 1999, 40, 793–795. |
|
Joint International Symposium on Molten |
|
45 |
P. Ludley, N. Karodia, Tetrahedron Lett. |
|
Salts, 6th ed., 1987, 979–990. |
||
|
|
2001, 42, 2011–2014. |
|
67 |
K. K. Laali, V. J. Gettwert, J. Org. Chem. |
46 |
F. Zulfiqar, T. Kitazume, Green Chem. |
|
2001, 66, 35–40. |
||
|
|
2000, 2, 137–139. |
|
68 |
Y. Patell, N. Winterton, K. R. Seddon, |
47 |
W. T. Ford, R. J. Hauri, D. J. Hart, |
|
World Patent WO 0037400, 2000. |
||
|
|
J. Org, Chem. 1973, 38, 3916–3918. |
69 |
D. S. Newman, T. H. Kinstle, G. Tham- |
|
48 |
W. T. Ford, R. J. Hauri, J. Am. Chem. |
|
bo, Proceedings of the Joint International |
||
|
|
Soc. 1973, 95, 7381–7391. |
|
Symposium on Molten Salts, 6th ed., |
|
49 |
W. T. Ford, J. Org, Chem. 1973, 38, |
|
1987, 991–1001. |
||
|
|
3614–3615. |
|
70 |
D. S. Newman, T. H. Kinstle, G. Tham- |
50 |
M. Badri, J.-J. Brunet, Tetrahedron Lett. |
|
bo, J. Electrochem. Soc. 1987, 134, C512. |
||
|
|
1992, 33, 4435–4438. |
|
71 |
D. S. Newman, R. E. Winans, R. L. |
51 |
M. J. Earle, P. B. McCormac, K. R. |
|
McBeth, J. Electrochem. Soc. 1984, 131, |
||
|
|
Seddon, Chem. Commun. 1998, |
|
1079–1083. |
|
|
|
2245–2246. |
|
72 |
F. Zulfiqar, T. Kitazume, Green Chem. |
52 |
C. Wheeler, K. N. West, C. L. Liotta, |
|
2000, 2, 296–298. |
||
|
|
C. A. Eckert, Chem. Commun. 2001, |
73 |
S.-gi Lee, J. H. Park, J. Kang, J. K. Lee, |
|
|
|
887–888. |
|
|
Chem. Commun. 2001, 1698–1699. |
53 |
M. J. Earle, P. B. McCormac, K. R. |
74 |
G. A. Olah, Friedel–Crafts and Related |
||
|
|
Seddon, Green Chem. 2000, 2, |
|
Reactions, Interscience , New York, |
|
|
|
261–262. |
|
|
1963. |
54 |
C. M. Gordon, A. McClusky, Chem. |
75 |
G. A. Olah, Friedel Crafts Chemistry, |
||
|
|
Commun. 1999, 143–144. |
|
Wiley–Interscience, New York, 1973. |
|
55 |
T. Kitazume, K. Kasai, Green Chem. |
76 |
G. Baddeley, R. Williamson, J. Chem. |
||
|
|
2001, 3, 30–32. |
|
|
Soc. 1953, 2120–2123. |
56 |
A. McCluskey, J. Garner, D. J. Young, |
77 |
G. Baddeley, G. Holt, S. M. Makar, |
||
|
|
S. Caballero, Tetrahedron Lett, 2000, |
|
M. G. Ivinson, J. Chem. Soc. 1952, |
|
|
|
41, 8147–8151. |
|
|
3605–3607. |
57 |
G. W. Kabalka, R. R. Malladi, Chem. |
78 |
W. L. Mendelson, C. B. Spainhour, |
||
|
|
Commun. 2000, 2191. |
|
|
S. S. Jones, B. L. Lamb, K. L. Wert, |
58 |
T. Kitazume, F. Zulfiqar, G. Tanaka, |
|
Tetrahedron Lett. 1980, 21, 1393–1396. |
||
|
|
Green Chem., 2000, 2, 133–136. |
79 |
J. A. Boon, J. A. Levisky, J. L. Pflug, |
|
59 |
P. P. Peng, Y. Q. Deng, |
Tetrahedron |
|
J. S. Wilkes, J. Org. Chem, 1986, 51, |
|
|
|
Lett. 2001, 42, 403–405. |
|
|
480–483. |
60 |
T. Kitazume, G. Tanaka, |
J. Fluorine. |
80 |
B. J. Piersma, M. Merchant in Proceed- |
|
|
|
Chem. 2000, 106, 211–215. |
|
ings of the 7th International Symposium |
|
61 |
D. W. Morrison, D. C. Forbes, J. H. |
|
on Molten Salts (C. L. Hussey, S. N. |
||
|
|
Davis Jr., Tetrahedron Lett. 2001, 42, |
|
Flengas, J. S. Wilkes, Y. Ito eds.), The |
|
|
|
6053–6055. |
|
|
Electrochemical Society, Pennington, |
|
|
|
|
|
1990, 805–821. |
|
|
5.2 Transition Metal Catalysis in Ionic Liquids |
213 |
|
|
|
|
|
|
81 |
P. K. G. Hodgson, M. L. M. Morgan, |
93 |
G. C. Misra, L. M. Pande, G. C. Joshi, |
|
|
B. Ellis, A. A. K. Abdul-Sada, M. P. |
|
A. K. Misra, Aust. J. Chem. 1972, 25, |
|
|
Atkins, K. R. Seddon, US Patent, US |
|
1579–1581. |
|
|
5994602, 1999. |
94 |
S. Wagatsuma, H. Higuchi, T. Ito, T. |
|
82 |
P. Wasserscheid, B. Ellis, H. Fabi- |
|
Nakano, Y. Naoi, K. Sakai, T. Matsui, |
|
|
enne, World Patent, WO 0041809, |
|
Y. Takahashi, A. Nishi, S. Sano, Org. |
|
|
2000. |
|
Prep. Proced. Int. 1973, 5, 65–70. |
|
83 |
C. C. Greco, S. Fawzy, S. Lieh-Jiun, |
95 |
C. J. Adams, M. J. Earle, G. Roberts, |
|
|
US Patent, US 5824832, 1998. |
|
K. R. Seddon, Chem. Commun. 1998, |
|
84 |
W. Keim, W. Korth, P. Wasserscheid, |
|
2097–2098. |
|
|
World Patent, WO 0016902, 2000. |
96 |
P. N. Davey, M. J. Earle, C. P. New- |
|
85 |
C. E. Song, W. H. Shim, E. J. Roh, |
|
man, K. R. Seddon, World Patent WO |
|
|
J. H. Choi, Chem. Commun. 2000, |
|
99 19288, 1999. |
|
|
1695–1696. |
97 |
M. H. Valkenberg, C. deCastro, W. F. |
|
86 |
M. J. Earle, unpublished results, 1998. |
|
Hölderich, App. Catal. A 2001, 215, |
|
87 |
C. P. DeCastro, E. Sauvage, M. H. |
|
185–190. |
|
|
Valkenberg, W. F. Hölderich, World |
98 |
G. L. Rebeiro, B. M. Khadilkar, Syn. |
|
|
Patent, WO 0132308, 2001. |
|
Commun. 2000, 30, 1605–1608. |
|
88 |
C. P. DeCastro, E. Sauvage, M. H. |
99 |
C. J. Adams, M. J. Earle, K. R. Seddon, |
|
|
Valkenberg, W. F. Hölderich, J. Catal. |
|
Green Chem. 2000, 2, 21–24. |
|
|
2000, 196, 86–94. |
100 |
R. W. J. Westerhout, J. A. M. Kuipers, |
|
89 |
J. L. Scott, D. R. MacFarlan, C. L. Ras- |
|
W. P. M. van Swaaij, Ind. Eng. Chem. |
|
|
ton, C. M. Teoh, Green Chem. 2000, 2, |
|
Res. 1998, 37, 841–847. |
|
|
123–126. |
101 |
P. N. Barnes, K. A. Grant, K. J. Green, |
|
90 |
H. Raudnitz, G. Laube, Ber. 1929, 62, |
|
N. D. Lever, World Patent WO |
|
|
509. |
|
0040673, 2000. |
|
91 |
R. Scholl, K. Meyer, J. Donat, Chem. |
102 |
C. J. Adams, M. J. Earle, J. Hamill, |
|
|
Ber. 1937, 70, 2180–2189. |
|
C. M. Lok, G. Roberts, K. R. Seddon, |
|
92 |
D. B. Bruce, A. J. S. Sorrie, R. H. |
|
World Patent WO 9807680, 1998. |
|
|
Thomson, J. Chem. Soc. 1953, |
103 |
C. J. Adams, M. J. Earle, J. Hamill, |
|
|
2403–2408. |
|
C. M. Lok, G. Roberts, K. R. Seddon, |
|
|
|
|
World Patent WO 9807679, 1998. |
|
5.2
Transition Metal Catalysis in Ionic Liquids
Peter Wasserscheid
Many transition metal complexes dissolve readily in ionic liquids, which enables their use as solvents for transition metal catalysis. Sufficient solubility for a wide range of catalyst complexes is an obvious, but not trivial, prerequisite for a versatile solvent for homogenous catalysis. Some of the other approaches to the replacement of traditional volatile organic solvents by “greener” alternatives in transition metal catalysis, namely the use of supercritical CO2 or perfluorinated solvents, very often suffer from low catalyst solubility. This limitation is usually overcome by use of special ligand systems, which have to be synthesized prior to the catalytic reaction.
In the case of ionic liquids, special ligand design is usually not necessary to obtain catalyst complexes dissolved in the ionic liquid in sufficiently high concentrations.

214 Peter Wasserscheid
However, it should be mentioned that the dissolution process of a solid, crystalline complex in an (often relatively viscous) ionic liquid can sometimes be slow. This is due to restricted mass transfer and can be speeded up either by increasing the exchange surface (ultrasonic bath) or by reducing the ionic liquid’s viscosity. The latter is easily achieved by addition of small amounts of a volatile organic solvent that dissolves both the catalyst complex and the ionic liquid. As soon as the solution is homogeneous, the volatile solvent is then removed in vacuo.
Since no special ligand design is usually required to dissolve transition metal complexes in ionic liquids, the application of ionic ligands can be an extremely useful tool with which to immobilize the catalyst in the ionic medium. In applications in which the ionic catalyst layer is intensively extracted with a non-miscible solvent (i.e., under the conditions of biphasic catalysis or during product recovery by extraction) it is important to ensure that the amount of catalyst washed from the ionic liquid is extremely low. Full immobilization of the (often quite expensive) transition metal catalyst, combined with the possibility of recycling it, is usually a crucial criterion for the large-scale use of homogeneous catalysis (for more details see Section 5.3.5).
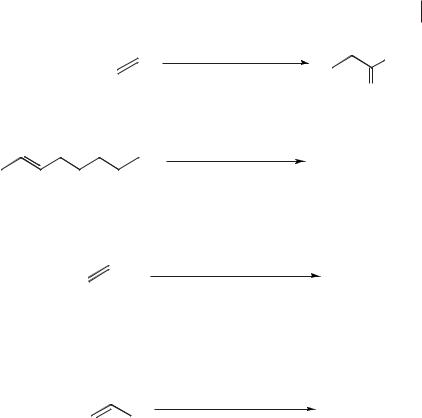
The first example of homogeneous transition metal catalysis in an ionic liquid was the platinum-catalyzed hydroformylation of ethene in tetraethylammonium trichlorostannate (mp. 78 °C), described by Parshall in 1972 (Scheme 5.2-1, a)) [1]. In 1987, Knifton reported the rutheniumand cobalt-catalyzed hydroformylation of internal and terminal alkenes in molten [Bu4P]Br, a salt that falls under the now accepted definition for an ionic liquid (see Scheme 5.2-1, b)) [2]. The first applications of room-temperature ionic liquids in homogeneous transition metal catalysis were described in 1990 by Chauvin et al. and by Wilkes et al.. Wilkes et al. used weekly acidic chloroaluminate melts and studied ethylene polymerization in them with Ziegler–Natta catalysts (Scheme 5.2-1, c)) [3]. Chauvin’s group dissolved nickel catalysts in weakly acidic chloroaluminate melts and investigated the resulting ionic catalyst solutions for the dimerization of propene (Scheme 5.2-1, d)) [4].
The potential of ionic liquids as novel media for transition metal catalysis received a substantial boost from the work of Wilkes’ group, when in 1992 they described the synthesis of non-chloroaluminate, room-temperature liquid systems with significantly enhanced stability to hydrolysis, such as low-melting tetrafluoroborate melts [5]. In contrast to the chloroaluminate ionic liquids, these “second generation ionic liquids” offer high tolerance to functional groups, which opens up a much larger range of applications, especially for transition metal catalysis. The first successful catalytic reactions in tetrafluoroborate ion-based ionic liquids included the rhodium-catalyzed hydrogenation and hydroformylation of olefins [6]. Nowadays, the tetrafluoroborate and the (published slightly later [7]) hexafluorophosphate ionic liquids are among the “workhorses” for transition metal catalysis in ionic liquids. They – like some other ionic liquids with weakly coordinating anions – combine the properties of relatively polar yet non-coordinating solvents. This special combination makes them extremely suitable solvents for reactions involving electrophilic catalysts [8]. Moreover, these ionic liquids are now widely commercially available [9], so research groups and companies focussing on catalyt-
5.2 Transition Metal Catalysis in Ionic Liquids 215
a) Parshall (1972):
PtCl2
[NEt4][SnCl3] 90°C, 400bar
b) Knifton (1987):
RuO2
[PBu4]Br
180°C, 83 bar CO/H2 (1:2)
c) Wilkes et al. (1990):
Cp2TiCl2
[EMIM]Cl/AlCl3 (Al molar fraction= 0.53)
25°C, 1 bar ethene pressure
d) Chauvin et al. (1990):
NiCl2(PiPr3)2
[BMIM]Cl/AlEtCl2
(Al molar fraction = 0.7) -15°C
Scheme 5.2-1: Early examples of transition metal catalysis in ionic liquids.
H
O
nonanol isomers
PE
C6-dimers
ic applications do not necessarily have to go through all of the synthetic work themselves (for the synthesis of ionic liquids, and especially for the quality requirements related to their applications as solvents in homogeneous catalysis, see Chapter 2).
However, a number of limitations are still evident when tetrafluoroborate and hexafluorophosphate ionic liquids are used in homogeneous catalysis. The major aspect is that these anions are still relatively sensitive to hydrolysis. The tendency to anion hydrolysis is of course much less pronounced than that of the chloroaluminate melts, but it still occurs and this has major consequences for their use in transition metal catalysis. For example, the [PF6]– anion of 1-butyl-3-methylimida- zolium ([BMIM]) hexafluorophosphate was found (in the author’s laboratories) to hydrolyze completely after addition of excess water when the sample was kept for 8 h at 100 °C. Gaseous HF and phosphoric acid were formed. Under the same conditions, only small amounts of the tetrafluoroborate ion of [BMIM][BF4] was converted into HF and boric acid [10]. The hydrolytic formation of HF from the anion of the ionic liquid under the reaction conditions causes the following problems with