

Wasserscheid P., Welton T. - Ionic Liquids in Synthesis (2002)(en)
.pdf286 Peter Wasserscheid
In the biphasic batch reaction the best reaction conditions were found for the system [EMIM][(CF3SO2)2N]/compressed CO2. It was found that increasing the partial pressure of ethylene and decreasing the temperature helped to suppress the concurrent side reactions (isomerization and oligomerization), 58 % conversion of styrene (styrene/Ni = 1000/1) being achieved after 1 h under 40 bar of ethylene at 0 °C with 3-phenyl-1-butene being detected as the only product and with a 71 % ee of the R isomer.
However, attempts to reuse the ionic catalyst solution in consecutive batches failed. While the products could readily be isolated after the reaction by extraction with scCO2, the active nickel species deactivated rapidly within three to four batchwise cycles. The fact that no such deactivation was observed in later experiments with the continuous flow apparatus described below (see Figure 5.4-2) clearly indicate the deactivation of the chiral Ni-catalyst being mainly related to the instability of the active species in the absence of substrate.
In the continuous hydrovinylation experiments, the ionic catalyst solution was placed in the reactor R, where it was in intimate contact with the continuous reaction phase entering from the bottom (no stirring was used in these experiments). The reaction phase was made up in the mixer from a pulsed flow of ethylene and a continuous flow of styrene and compressed CO2.
|
|
DP |
|
|
PT |
P |
D |
|
|
|
|
|
C |
|
S |
|
MF |
|
|
|
C2H4 |
CO2 |
F |
CT |
||
|
M |
R |
|
|
MF |
Figure 5.4-2: Schematic view of the continuous flow apparatus used for the enantioselective hydrovinylation of styrene in the biphasic [EMIM][(CF3SO2)2N] system. The components are labeled (alphabetically) as follows: C: compressor, CT: cold trap, D: dosimeter, DP: depressurizer, F: flow-meter, M: mixer, MF: metal filter, P: HPLC pump, PT: pressure transducer and thermocouple, R: reactor, S: styrene.
5.4 Multiphasic Catalysis with Ionic Liquids in Combination with Compressed CO2 287
90 |
|
|
|
|
|
|
85 |
|
|
|
|
|
|
80 |
|
|
|
|
|
|
75 |
|
|
|
|
|
|
70 |
|
|
|
|
|
|
% 65 |
|
|
|
|
|
|
60 |
|
|
|
|
|
|
55 |
|
|
|
|
|
|
50 |
|
|
|
|
|
|
45 |
|
|
|
|
|
|
40 |
|
|
|
|
|
|
0 |
10 |
20 |
30 |
40 |
50 |
60 |
|
|
t |
/h |
|
|
|
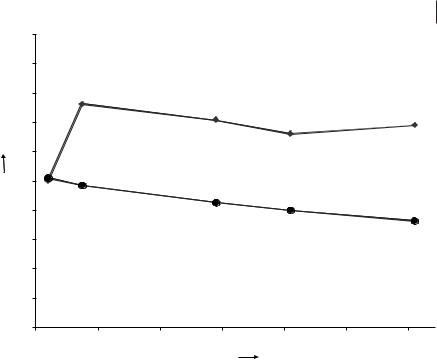
Lifetime study of Wilke’s catalyst in the hydrovinylation of styrene, activated and immobilized in the [EMIM][(CF3SO2)2N]/compressed CO2 system (● ee; ♦ conversion).
Figure 5.4-3 shows the results of a lifetime study for Wilke’s catalyst dissolved, activated, and immobilized in the [EMIM][(CF3SO2)2N]/compressed CO2 system. Over a period of more than 61 h, the active catalyst showed remarkably stable activity while the enantioselectivity dropped only slightly. These results clearly indicate
– at least for the hydrovinylation of styrene with Wilke’s catalyst – that an ionic liquid catalyst solution can show excellent catalytic performance in continuous product extraction with compressed CO2.
5.4.6
Concluding Remarks and Outlook
The combination of ionic liquids and compressed CO2 – at opposite extremes of the volatility and polarity scales – offers a new and intriguing immobilization technique for homogeneous catalysis.
In comparison with catalytic reactions in compressed CO2 alone, many transition metal complexes are much more soluble in ionic liquids without the need for special ligands. Moreover, the ionic liquid catalyst phase provides the potential to activate and tune the organometallic catalyst. Furthermore, product separation from the catalyst is now possible without exposure of the catalyst to changes of temperature, pressure, or substrate concentration.


Ionic Liquids in Synthesis. Edited by Peter Wasserscheid, Thomas Welton Copyright © 2002 Wiley-VCH Verlag GmbH & Co. KGaA
ISBNs: 3-527-30515-7 (Hardback); 3-527-600701 -Kolumnentitel1 (Electronic) 289
6
Inorganic Synthesis
Frank Endres and Tom Welton
6.1
Directed Inorganic and Organometallic Synthesis
Tom Welton
Although a great deal of excitement has surrounded the use of ionic liquids as solvents for organic synthesis, the rational synthesis of inorganic and organometallic compounds in ionic liquids has remained largely unexplored.
6.1.1
Coordination Compounds
Some halogenometalate species have been observed to have formed spontaneously during spectroelectrochemical studies in ionic liquids. For example, [MoCl6]2– (which is hydrolyzed in water, is coordinated by solvent in polar solvents, and has salts that are insoluble in non-polar solvents) can only be observed in basic {X(AlCl3) < 0.5} chloroaluminate ionic liquids [1]. However, this work has been directed at the measurement of electrochemical data, rather than exploitation of the ionic liquids as solvents for synthesis [2]. It has been shown that the tetrachloroaluminate ion will act as a bidentate ligand in acidic {X(AlCl3) > 0.5} chloroaluminate ionic liquids, forming [M(AlCl4)3]– ions [3]. This was also the result of the spontaneous formation of the complexes, rather than a deliberate attempt to synthesize them.
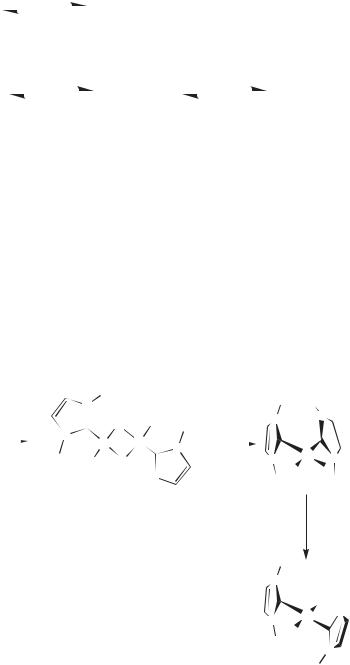
The only reports of directed synthesis of coordination complexes in ionic liquids are from oxo-exchange chemistry. Exposure of chloroaluminate ionic liquids to water results in the formation of a variety of aluminium oxoand hydroxo-contain- ing species [4]. Dissolution of metals more oxophilic than aluminium will generate metal oxohalide species. Hussey et al. have used phosgene (COCl2) to deoxochlorinate [NbOCl5]2– (Scheme 6.1-1) [5].
290 |
Tom Welton |
|
|
|
|
|
|
|
||||||
|
|
|
|
|
COCl2 |
|
Scheme 6.1-1: |
Nb(V) oxo-exchange |
||||||
|
|
|
|
|
|
|||||||||
|
[NbOCl5]2- |
|
|
|
|
[NbCl6]- chemistry in a basic [EMIM]Cl/AlCl3 |
||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
||||||||||
|
|
|
|
|
O2- |
|
ionic liquid. |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
[VO2Cl2]- |
|
triphosgene |
|
|
triphosgene |
||||||||
|
|
|
|
|
[VOCl4]2- |
|
|
|
|
[VCl6]3- |
||||
|
|
|
|
|
|
|
|
|
||||||
|
PhIO |
|
O2 |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
||||
Scheme 6.1-2: Vanadium oxo-exchange chemistry in a basic [EMIM]Cl/AlCl3 ionic liquid
Triphosgene (bis(trichloromethyl)carbonate) has been used to deoxochlorinate [VOCl4]2– to [VCl6]3– and [VO2Cl2]– to [VOCl4]2– [6]. In both these cases the deoxochlorination was accompanied by spontaneous reduction of the initial products (Scheme 6.1-2).
6.1.2
Organometallic Compounds
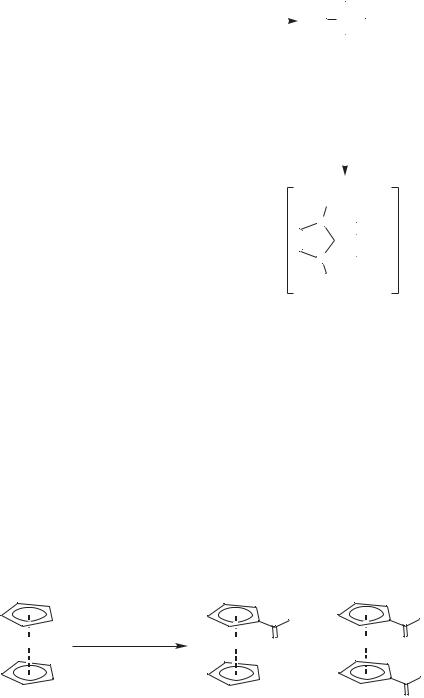
With the enthusiasm currently being generated by the (so-called) stable carbenes (imidazolylidenes) [7], it is surprising that there are few reports of imidazoliumbased ionic liquids being used to prepare metal imidazolylidene complexes. Xiao et al. have prepared bis(imidazolylidene)palladium(II) dibromide in [BMIM]Br [8]. All four possible conformers are formed, as shown in Scheme 6.1-3.
|
|
|
Me |
|
|
|
|
Me |
Bu |
|
|
|
|
N |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Br |
Br |
|
|
N |
|
N |
|
|
|
|
|
|
|
|
|||
|
[bmim]Br |
N |
|
Bu |
|
|
||||
Pd(OAc)2 |
Pd |
|
|
|
|
|
|
|||
|
|
Pd |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||||
|
Bu |
|
N |
Pd |
|
|||||
|
|
|
|
|
|
|||||
|
|
Br |
Br |
|
|
|
N |
N |
||
|
|
|
|
|
|
|||||
|
|
|
|
|
N |
|
|
Br |
|
Br |
|
|
|
|
Me |
|
|
Bu |
|
Me |
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
|
|
N |
|
|
|
|
|
Bu |
|
|
Br |
|
|
N |
Pd |
N |
|
|
|
||
|
|
|
|
Br |
|
|
|
Bu |
N |
|
|
|
|
|
|
|
Me |
|
|
Scheme 6.1-3: The formation of bis(1-butyl-3-methylimidazolylidene)palladium(II) dibromide in [BMIM]Br
|
|
6.1 Directed Inorganic and Organometallic Synthesis |
291 |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PdX2 |
|
|
NaX |
|
|
|
|
|
|
PPh3 |
||||||||
|
|
|
||||||||||||||||
|
|
|
|
X |
|
|
|
|
|
|
|
|
X |
|||||
|
|
|
Pd |
|
|
|
|
|||||||||||
or |
+ PPh3 |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|||||||||||||
|
[BMIM][BF4] |
|
|
|
|
|
|
|
|
|
||||||||
Pd(OAc)2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
PPh3 |
||||||||||
|
|
|
|
31P{1H} = 24.2 ppm (Cl) |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
22.6 ppm (Br) |
||||||
|
|
|
|
|
|
|
|
|
|
Na2CO3 |
||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|||||
|
|
|
|
|
|
Me |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
N |
|
|
PPh3 |
|||||||||
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
Pd |
|
X |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
PPh3 |
||||||||||||||
|
|
|
|
|
|
Bu |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
31P{1H} = 22.9 ppm (Cl) |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
21.8 ppm (Br) |
||||||
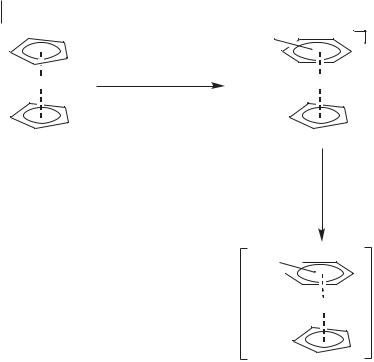
Scheme 6.1-4: The formation of (1-butyl-3-methylimidazolylidene)bis(triphenylphos- phine)palladium(II) chloride in [BMIM][BF4].
In the presence of triphenylphosphine and four equivalents of chloride, (1-butyl- 3-methylimidazolylidene)bis(triphenylphosphine)palladium(II) chloride is formed (Scheme 6.1-4).
Singer and co-workers have investigated the acylation reactions of ferrocene in ionic liquids made from mixtures of [EMIM]I and aluminium(III) chloride (Scheme 6.1-5) [9, 10]. The ionic liquid acts both as solvent and as source of the Friedel–Crafts catalyst. In mildly acidic {X(AlCl3) > 0.5} [EMIM]I/AlCl3, the monoacetylated ferrocene was obtained as the major product. In strongly acidic [EMIM]I/AlCl3 {X(AlCl3) = 0.67} the diacylated ferrocene was the major product. Also, when R = alkyl, the diacetylated product was usually the major product, but for R = Ph, the monoacetylated product was favored.
|
|
|
R |
|
R |
Fe |
RCOCl or (RCO)2O |
O |
+ |
Fe |
O |
Fe |
|||||
|
[EMIM]I-AlCl3 |
|
|
|
|
|
|
|
|
|
R |
Scheme 6.1-5: |
The acylation of ferrocene in [EMIM]I/AlCl3 [9, 10]. |
|
|
O |
|
292 Tom Welton
|
Ar |
+ |
|
|
|
Fe |
80 oC / 8 hours |
Fe |
|
acidic ionic liquid aluminium powder arene [bmim][HCl2]
H2O
NH4PF6
Ar 
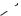
Fe [PF6]
Scheme 6.1-6: Arene exchange reactions of ferrocene in [BMIM]Cl/AlCl3.
In another study relying on chloroaluminate chemistry, the Fisher–Hafner-type ligand-exchange reactions of ferrocene were investigated (Scheme 6.1-6) [11]. Again, the acidic ionic liquids acted as combinations of solvent and catalyst. In these reactions it was necessary to add [BMIM][HCl2] as a proton source, to generate the cyclopentadiene leaving group.
The strong halide-abstracting properties of acidic {X(AlCl3) = 0.67} [BMIM]Cl/AlCl3 have been used for the synthesis of the “piano stool” complexes [Mn(CO)3(η6-arene)]+ (Scheme 6.1-7) [12].
In all of the above cases the products were isolated by the destruction of the chloroaluminate ionic liquids by addition to water.
6.1.3
Other Reactions
The only other report of the use of an ionic liquid to prepare an inorganic material is that of the formation of a silica aerogel in [EMIM][(CF3SO2)2N] [13]. Formic acid was added to tetramethylorthosilicate in the ionic liquid, yielding a gel that cured over a period of three weeks (Scheme 6.1-8). Here, it was the nonvolatile nature of the ionic liquid, preventing the loss of solvent during the curing process, that was exploited. The ionic liquid was retrieved from the aerogel by extraction with acetonitrile.
6.1 Directed Inorganic and Organometallic Synthesis 293
|
Ar |
+ |
|
80 oC / 8 hours |
|
|
|
Mn(CO)5Br |
|
Mn |
|
acidic [BMIM]Cl-AlCl3 |
|
|
|
arene |
OC |
CO |
|
|
|
CO |
|
|
|
H2O |
|
|
|
NH4PF6 |
|
|
Ar |
|
|
|
|
Mn |
[PF6] |
|
OC |
CO |
|
|
|
CO |
|
Scheme 6.1-7: The synthesis of [Mn(CO)3(η6-arene)]+ “piano stool” complexes in [BMIM]Cl/AlCl3.
2 HC(O)OH + (CH3O)4Si  SiO2 + 2 CH3OH + 2 HC(O)OCH3 [EMIM][(CF3SO2)2N]
SiO2 + 2 CH3OH + 2 HC(O)OCH3 [EMIM][(CF3SO2)2N]
Scheme 6.1-8: The formation of a SiO2 aerogel in [EMIM][(CF3SO2)2N].
6.1.4
Outlook
There is no doubt that inorganic and organometallic synthesis in ionic liquids is lagging behind organic synthesis. This is not due to any lack of importance. If, for instance, ionic liquids are to find use in biphasic catalysis, a point will arrive at which the ionic liquid layer can no longer be recycled. It will only be through an understanding of the chemistry of the dissolved catalysts, deliberately prepared to be difficult to remove, that they will be transformable into materials capable of being extracted from the ionic liquids.
Ionic liquids hold as much promise for inorganic and organometallic synthesis as they do for organic synthesis. Their lack of vapor pressure has already been exploited [13], as have their interesting solubility properties. The field can only be expected to accelerate from its slow beginnings.

294 Frank Endres
References
1 |
T. B. Scheffler, C. L. Hussey, K. R. |
8 |
L. Xu, W. Chen, and J. Xiao, |
|
Seddon, C. M. Kear and P. D. |
|
Organometallics 2000, 19, 1123. |
|
Armitage, Inorg. Chem. 1984, 23, |
9 |
J. K. D. Surette, L. Green, and R. D. |
|
1926. |
|
Singer, Chem. Commun. 1996, 2753. |
2 |
C. L. Hussey, Pure Appl. Chem. 1988, |
10 |
A. Stark, B. L. MacLean, and R. D. |
|
60, 1763. |
|
Singer, J. Chem. Soc., Dalton Trans. |
3 |
A. J. Dent, K. R. Seddon and T. Wel- |
|
1999, 63. |
|
ton, J. Chem. Soc., Chem. Commun. |
11 |
Paul J. Dyson, Martin C. Grossel, |
|
1990, 315. |
|
N. Srinivasan, T. Vine, T. Welton, |
4 |
T. Welton, Chem. Rev. 1999, 99, 2071. |
|
D. J. Williams, A. J. P. White, and |
5 |
I. W. Sun, E. H. Ward and C. L. |
|
T. Zigras, J. Chem. Soc., Dalton Trans. |
|
Hussey, Inorg. Chem. 1987, 26, 4309. |
|
1997, 3465. |
6 |
A. J. Dent, A. Lees, R. J. Lewis, and |
12 |
D. Crofts, P. J. Dyson, K. M. Sander- |
|
T. Welton, J. Chem. Soc., Dalton |
|
son, N. Srinivasan, and T. Welton, |
|
Trans. 1996, 2787. |
|
J. Organomet. Chem. 1999, 573, 292 |
7 |
(a) W. A. Herrmann and C. Kocher, |
13 |
S. Dai, Y. H. Ju, H. J. Gao, J. S. Lin, |
|
Angew. Chem. Int. Ed. 1997, 36, 2163; |
|
S. J. Pennycook, and C. E. Barnes, |
|
(b) D. Bourissou, O Guerret, F. P. |
|
Chem. Commun. 2000, 243. |
|
Gabbai and G. Bertrand, Chem. Rev. |
|
|
|
2000, 100, 39. |
|
|
6.2
Making of Inorganic Materials by Electrochemical Methods
Frank Endres
6.2.1
Electrodeposition of Metals and Semiconductors
6.2.1.1General considerations
Electrodeposition is one of the main fields in electrochemistry, both in industrial processes and in basic research. In principal, all metals and semiconductors can be obtained by electrolysis of the respective salts in aqueous or organic solutions and molten salts, respectively. As well as electrowinning of the elements, electrocoating of materials for corrosion protection is an important field in industry and in basic research. With the help of the Scanning Tunneling Microscope, a great deal of work on the nanometer scale has been done over the past 15 years. Insight into how the initial stages of metal deposition influence the bulk growth has been obtained. Furthermore, the role of brighteners, added to solutions to make shining deposits, can now be understood; they seem to adsorb at growing clusters and force the metal to grow layer-by-layer instead of in the form of clusters [1]. Aqueous solutions, however, are unsuitable for the electrodeposition of less noble elements because of their limited electrochemical windows. For light, refractory, and rare earth metals, water

6.2 Making of Inorganic Materials by Electrochemical Methods 295
fails as a solvent because hydrogen evolves long before deposition of the metal. Ionic liquids are ideal solvents for such purposes though, because they have – depending on their compositions – wide electrochemical windows combined with good solubilities of most metal salts and semiconductor compounds [2]. Many technical processes, such as the electrowinning of the rare earth and refractory metals Mg, Al, and several others, are performed in high-temperature molten salts. These systems are highly corrosive and sometimes make it difficult to find materials that will withstand chemical attack by the melts. The design of electrochemical cells for low-melting ionic liquids, in contrast, is much easier. These combine, more or less, the advantages of classical molten salts and those of aqueous media. Thanks to their wide electrochemical windows, several metals and alloys conventionally accessible from high-temperature molten salts – such as Al and its alloys, La, etc. – can also be deposited at room temperature. Furthermore, metals obtainable from aqueous media can in most cases also be deposited from ionic liquids, often with superior quality since hydrogen evolution does not occur. Pd is a good example, since deposits from aqueous solutions can contain varying amounts of hydrogen, which can make the deposits rather brittle. However, shining, even, nanosized Pd deposits can easily be obtained from ionic liquids. These features and their good ionic conductivities of between 10–3 and 10–2 (Ωcm)–1 make the ionic liquids interesting solvents for electrodeposition.
Section 6.2.1 offers literature data on the electrodeposition of metals and semiconductors from ionic liquids and briefly introduces basic considerations for electrochemical experiments. Section 6.2.2 describes new results from investigations of process at the electrode/ionic liquids interface. This part includes a short introduction to in situ Scanning Tunneling Microscopy.
6.2.1.2Electrochemical equipment
Any redox couple has a defined electrode potential on the electrochemical potential scale. In aqueous solutions, for example, a silver wire immersed into a solution containing Ag+ ions with the activity 1 has a value of +799 mV vs. the normal hydrogen electrode. At more positive potentials a Ag electrode will dissolve, at more negative values Ag will deposit from the ions. If one applies a certain voltage between two electrodes, an electrochemical reaction may occur, depending on the applied voltage. If one wants to know the processes involved, for example during electrodeposition, it is necessary to know the electrode potential relative to a reference electrode exactly. This can be measured by means of a third electrode immersed in the solution. This is the three-electrode setup, with the working electrode of interest (WE), the reference electrode (RE), and the counter-electrode (CE). However, any current I(EC) that flows through the cell will influence the electrode potentials, so a stable value would be hard to obtain. A potentiostat allows precise control of the potential of the working electrode with respect to a reference electrode. It always applies the desired value U(setpoint) to the working electrode, usually responding to changes within microseconds. A simplified setup based on an operational amplifier (OPA), where the working electrode is connected to ground, is presented in Figure 6.2-1.