

17. Метод мо Хюккеля
Этот не-ССП метод, предложенный для углеводородов основан на нескольких очень сильных приближениях:
-электронное приближение; считают, что АО образуют ортонормальный базис,
т.е. S = .
2) Межэлектронными взаимодействиями (т.е. всеми двухэлектронными кулоновскими и обменными интегралами) пренебрегают. Из-за этого решение уравнений метода не требует итераций и проводится в один шаг.
Матричные элементы оператора Фока оцениваются на основании эмпирической информации и являются фиксированными:
h = ,
h = k . (78)
= I называется кулоновским интегралом (не путать с двухэлектронными кулоновскими интегралами AB) и принимается равным потенциалу ионизации электрона на орбитали в свободном атоме.
4) Считают, что =0, если АО и не принадлежат ковалентно связанным атомам.
После этих приближений уравнения Рутана-Хюккеля имеют вид
![]() (79)
(79)
Они имеют ненулевые решения при равенстве нулю детерминанта
h - i = 0. (80)
Полная энергия в методе Хюккеля есть сумма орбитальных энергий
![]() ,
n=0, 1 или 2, (81)
,
n=0, 1 или 2, (81)
где n – число электронов на МО.
Решим уравнения Хюккеля для молекулы этилена С2Н4, имеющей 2 -электрона (свяжем их с (рz) -АО атомов углерода, направленными перпендикулярно плоскости молекулы). Величины интегралов следующие: С = - 11.0 эВ, СС - 2.4 эВ. Детерминант (83) имеет вид
![]() (82)
(82)
х=(С - )/СС. Раскрывая определитель, имеем х2 –1=0 и х = 1, откуда
1= С +СС , 2 =С - СС. (83)
Система уравнений имеет вид
с1 х + с2 = 0
с1 + с2 х = 0, (84)
Подставим
х =
1 в (84). При х= -1 получим с1
= с2 .
Используя далее условие нормировки
волновой функции этилена с12
+ с22
= 1, получаем с1
= с2 =
![]() .
Таким образом, одна из-МО
этилена имеет вид
.
Таким образом, одна из-МО
этилена имеет вид
1
=
![]() (1
+2). (85)
(1
+2). (85)
При х = 1 имеем с1 = - с2 и, повторяя рассуждения, получаем другую -МО:
2
=
![]() (1
- 2). (86)
(1
- 2). (86)
Так как СС 0, то 1 2 причем, 1 - 2 = 2СС. Это означает, что МО 1 более энергетически стабильна.
18. Расширенный метод Хюккеля
Хоффман сохранил схему Хюккеля, но включил в рассмотрение все валентные (а не только ) орбитали и явно учел интегралы перекрывания - расширенный метод Хюккеля (РМХ).
Электронная энергия молекулы с закрытой оболочкой в методе РМХ является удвоенной суммой энергий занятых МО и описывается выражением
Е= 2( с2i h + сi сi h), (88)
(члены, ответственные за межэлектронное и межъядерное взаимодействие не учитываются). РМХ дает хорошие относительные оценки энергии в рядах соединений с равномерным распределением электронов. Гетероатомы нарушают равномерность электронного распределения и РМХ в соединениях с гетероатомами часто непригоден. Тем не менее, расширенный метод Хюккеля дает зачастую лучшие результаты, чем его простой аналог, при изучении конформаций циклических молекул, барьеров внутреннего вращения и передает относительный порядок уровней энергии.
19. Расчет свойств молекул
Таблица 20
Сравнительная характеристика всевалентных полуэмпирических методов [2]
|
Метод (приближение) |
Параметризуемое свойство |
Хорошо воспроизводимые свойства |
Плохо воспроизводимые свойства |
|
CNDO/2 (NDO) |
Электронная плотность |
Дипольные моменты, длины связей, валентные углы, силовые константы, ЯМР корреляции |
Теплоты образования, потенциал ионизации, сродство к электрону, спектры |
|
CNDO/S (NDO) |
Спектр |
Спектр |
Теплоты образования, геометрия |
|
INDO (NDO) |
Спиновые плотности |
Спиновые плотности, константы СТВ, геометрия |
Теплоты образования, потенциалы ионизации, сродство к электрону, спектры |
|
MINDO/3 (NDO) |
Потенциал атом-атомного взаимодействия |
Теплоты образования, потенциалы ионизации. Длины связей |
Спектры, водородная связь |
|
MNDO (NDDO) |
Теплоты образования |
Теплоты образования, геометрия |
Спектры, водородная связь |
|
АМ1 (NDDO) |
Теплоты образования |
Теплоты образования, геометрия |
Спектры |
|
РМ3 (NDDO) |
Теплоты образования, параметры межмолекулярного взаимодействия |
Теплоты образования, геометрия, водородная связь, межмолекулярное взаимодействие |
Спектры |
а) Энергия - важнейшая характеристика молекул. Любой квантово-химический расчет основан на минимизации энергии системы N электронов и M ядер.
Типичные результаты неэмпирического расчета молекулы CH4 по программе GAMESS (базис 6-31G*).
1 атомная единица. = 2625.5 кДж/моль.
ENERGY COMPONENTS:
ONE ELECTRON ENERGY = -79.8636554561
NUCLEUS-ELECTRON POTENTIAL ENERGY = -120.0455090699
TWO ELECTRON ENERGY = 26.1468653703
ELECTRON-ELECTRON POTENTIAL ENERGY = 26.1468653703
NUCLEAR REPULSION ENERGY = 13.5150853014
NUCLEUS-NUCLEUS POTENTIAL ENERGY = 13.5150853014
TOTAL ENERGY = -40.2017047844
TOTAL POTENTIAL ENERGY = -80.3835583982
TOTAL KINETIC ENERGY = 40.1818536137
WAVEFUNCTION NORMALIZATION = 1.0000000000
VIRIAL RATIO (V/T) = 2.0004940332
NUCLEAR ENERGY = 13.5150853014
ELECTRONIC ENERGY = -53.7167900858
TOTAL ENERGY = -40.2017047844
Полная энергия есть сумма потенциальной энергии отталкивания ядер и электронной энергии.
ELECTRONIC ENERGY = ONE ELECTRON ENERGY + TWO ELECTRON ENERGY =
= -79.8636554561 + 26.1468653703 = -53.7167900858
TOTAL ENERGY= NUCLEAR REPULSION ENERGY+ELECTRONIC ENERGY
= 13.5150853014 - 53.7167900858= -40.2017047844
Для оценки теплоты образования чаще используются полуэмпирические методы. Рассмотрим такой расчет для молекулы СН2 . Для определения теплоты образования вначале рассчитывается энергия атомизации (полная и электронная энергия для атомов одинакова):
Eатом. = -Etotal(CH2) +EC + 2EH. (89)
Etotal(CH2) (MNDO) = –151.586368 эВ,
EC –120.500606 эВ и EH –11.906276 эВ,
Eатом. = 151.586368 –120.500606–2*11.906276 = 7.2731эВ =167.37 ккал/моль.
Теплота образования метилена вычисляется с использованием экспериментальных значений теплот образования атомов углерода (170.89 ккал/моль) и водорода (52.102 ккал/моль):
fН0 = fН0© + 2*fН0(H) - Eатом. = 170.89 + 2*52.102 - 167.37 = 107 ккал/моль.
Полученный результат отвечает «химической точности» расчета теплот образования. Наилучшие результаты обеспечиваются приближениями АМ1 и РМ3: число элементов, по которых эти методы параметризованы, растет с каждым годом.
б) Дипольные и квадрупольные моменты молекул характеризуют распределение заряда по молекуле и определяют энергию межмолекулярного взаимодействия.
Дипольный момент
![]() (90)
(90)
(![]() -электронная
плотность молекулы,Za
и Ra
–заряд и координата ядра)
в
концентрированном виде характеризует
полярность молекулы. Принимают, что
диполь направлен от центра тяжести
положительных зарядов к центру тяжести
отрицательных зарядов. Для электронейтральных
молекул момент не зависит от выбора
начала отсчета.
-электронная
плотность молекулы,Za
и Ra
–заряд и координата ядра)
в
концентрированном виде характеризует
полярность молекулы. Принимают, что
диполь направлен от центра тяжести
положительных зарядов к центру тяжести
отрицательных зарядов. Для электронейтральных
молекул момент не зависит от выбора
начала отсчета.
Абсолютные значения дипольных моментов могут быть определены методом молекулярных пучков, микроволновой спектроскопией, измерением комплексной диэлектрической проницаемости как функции частоты и температуры, ИК-спектроскопией и с помощью других методов. Однако, только квантово-химический расчет позволяет определить точное направление (или знак) дипольного момента.
Таблица 21
Дипольные моменты некоторых молекул (10-30 К м)
|
молекула |
расчет (6-31G**) |
эксперимент (измерения в газе или растворе) |
|
Вода H2O |
+7.29 |
6.186 |
|
Цианамид CH2N2 |
+16.26 |
13.3 – 15.1 |
|
Формамид CH3NO |
+14.18 |
12.4(2) |
|
Мочевина CH4NO |
+17.06 |
15.2(1) |
|
L-аланин |
+41.44 |
41.0 |
|
Урацил C4H4N2O2 |
+16.22 |
13.9(1) |
|
пара-нитропиридин-N-оксид C5H4N2O3 |
+1.00 |
2.3(1) |
Квадрупольный момент характеризует отклонение распределения заряда от сферического. Он играет особенно важную роль, если дипольный момент нейтральной молекулы равен нулю, характеризуя распределение электронной плотности и определяет электрическое поле вокруг молекулы.
Определение квадрупольного момента следующее:
![]() (91)
(91)
(![]() - символ Кронекера). Квадрупольный момент
есть симметричный тензор 2-ого ранга,
сумма его диагональных элементов равна
нулю. Для нейтральных молекул с нулевым
дипольным моментом квадрупольный момент
не зависит от выбора системы координат.
Положительный знак
- символ Кронекера). Квадрупольный момент
есть симметричный тензор 2-ого ранга,
сумма его диагональных элементов равна
нулю. Для нейтральных молекул с нулевым
дипольным моментом квадрупольный момент
не зависит от выбора системы координат.
Положительный знак![]() указывает на «вытянутое» вдоль оси z
распределение заряда, отрицательный –
на «сплющенное» вдоль z распределение.
указывает на «вытянутое» вдоль оси z
распределение заряда, отрицательный –
на «сплющенное» вдоль z распределение.
Квадрупольный момент удобно выразить через так называемые вторые моменты электронного распределения
![]() (92)
(92)
θxx=μxx-(1/2)(μyy+μzz),
θxy=(3/2)μxy.
Другие компоненты θαβ получаются простой перестановкой индексов.
Простой неэмпирический расчет вполне удовлетворительно предсказывает их величины. Это обстоятельство позволяет, в частности, вести направленный поиск новых материалов, обладающих высокими нелинейными оптическими свойствами. Обуславливающие их электронные поляризуемости зависят от вторых и третьих моментов ЭП молекул в основном состоянии. Поэтому задача сводится к поиску веществ без центра симметрии, в которых взаимная ориентация молекул с большими вторыми и третьими моментами будет обеспечивать высокие нелинейно-оптические характеристики.
Таблица 22
Квадрупольные моменты некоторых молекул (10-40 К м2)
|
молекула |
компонента |
расчет (6-31G**) |
эксперимент |
|
вода Н2O |
θxx θyy θzz |
+7.93 -7.59 -0.33 |
+8.77(7) -8.34(7) -0.34(10) |
|
формамид CH3NO |
θxx θyy θzz |
-4.44 +12.58 -8.14 |
-1.0(7) -11.3(13) -10.3(27) |
|
ацетилен C2H2 |
θzz |
+23.23 |
+20.1(6) |
|
этилен C2H4 |
θxx θyy θzz |
+4.99 -11.04 +6.05 |
+4.7 -12.0 +7.3(10) |
|
s-триазин C3H3N3 |
θzz |
+2.03 |
-2.8(31) |
|
имидазол C3H4N2 |
θxx θyy θzz |
-1.58 +17.43 -15.84 |
-3.1(9) +22.6(11) -19.6(18) |
Квадрупольные моменты даны относительно центров масс молекул. Координатные оси выбраны так, чтобы максимально учесть симметрию молекул: ось z направлена вдоль оси 2-го порядка для молекул с симметрией С2v или D2h и перпендикулярно молекулярной плоскости для остальных молекул.
в) Молекулярный электростатический потенциал (МЭП) определяется электронной плотностью (r) и зарядами ядер Za:
![]() . (93)
. (93)
М
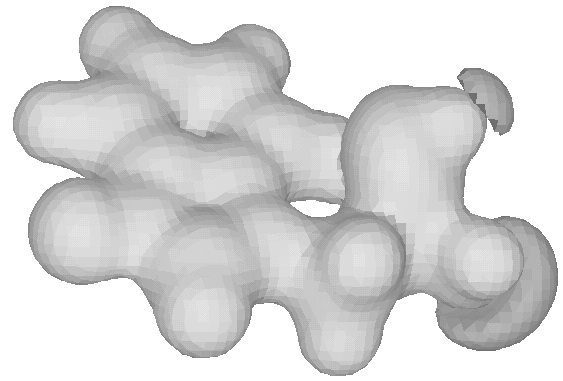

 ЭП
характеризует энергию электростатического
взаимодействия между молекулярным
распределением заряда и единичным
положительным бесконечно малым зарядом.
От МЭП зависят силы Гельмана-Фейнмана,
действующие на ядра молекулы при их
отклонениях от положения равновесия,
т.е. обуславливает равновесную
конфигурациюмолекулы. МЭП определяет
также активные центры взаимодействующих
молекул.
ЭП
характеризует энергию электростатического
взаимодействия между молекулярным
распределением заряда и единичным
положительным бесконечно малым зарядом.
От МЭП зависят силы Гельмана-Фейнмана,
действующие на ядра молекулы при их
отклонениях от положения равновесия,
т.е. обуславливает равновесную
конфигурациюмолекулы. МЭП определяет
также активные центры взаимодействующих
молекул.
Области отрицательного
потенциала
 а
б
а
б
Рис. 17. Молекула L-изомера незаменимой аминокислоты TRP (триптофана). (а) Карта распределения электростатического потенциала, (б) пространственная картина МЭП.
г) Расчет термодинамических функций. Неэмпирические расчеты с оптимизацией геометрии молекул дают возможность вычислить такие молекулярные характеристики, как частоты колебаний, вращательные моменты, и других величин, входящих в термодинамические функции. Наибольший интерес представляют, теплоемкость, внутренняя энергия, энтальпия, энтропия, энергия Гиббса, энергия Гельмгольца.
Полная внутренняя энергия одной молекулы есть сумма
Е = Еэл + Vяя + Тя. (94)
Электронная энергия молекулы Еэл в фиксированной равновесной геометрии (в приближении Борна-Оппенгеймера) описывается формулой (12), Vяя – энергия отталкивания ядер. Кинетическая энергия ядер Тя включает в себя вклады от поступательного, вращательного и колебательного движений:
Тя = Епост + Евр + Екол. (95)
Рассмотрим расчет этих величин..
Колебательная энергия. Минимум электронной энергии Еэл достигается при равенстве нулю производной энергии по координатам при неподвижных ядрах. Эти условия удовлетворяются оптимизацией геометрии молекулы. Раскладывая в ряд Тейлора Еэл как функцию координат ядер и выбрав начало координат в точке равновесного положения некоторого ядра, получим [2]:
![]() +…,
(96)
+…,
(96)
где
![]() - внутренние координаты (степени свободы
колебательного движения), число которых
равно (3N - 6). Для невырожденной матрицы
- внутренние координаты (степени свободы
колебательного движения), число которых
равно (3N - 6). Для невырожденной матрицы
 можно ввести новые координаты Q,
в которых отличными от нуля будут только
диагональные элементы
можно ввести новые координаты Q,
в которых отличными от нуля будут только
диагональные элементы
 .
Такие координаты называются нормальными,
а величина f
есть силовая постоянная колебания .
.
Такие координаты называются нормальными,
а величина f
есть силовая постоянная колебания .
Малые колебания ядер описываются уравнением Шредингера, в котором роль потенциальной энергии играет величина (1/2)fQ2 (задача о квантовом гармоническом осцилляторе). Возникает 3N-6 независимых уравнений, решения которых дают собственные значения энергии и собственные волновые функции колебательных состояний ядер. Для многоатомной молекулы энергия колебательного движения есть сумма энергий всех колебаний (без учета вырождения):
![]() ,
n
= 0, 1, 2, (97)
,
n
= 0, 1, 2, (97)
где
![]() - частоты
нормальных колебаний;
n - колебательные квантовые
числа,
h–
постоянная Планка, М
- приведенная масса ядра.
Нижний уровень энергии колебательного
движения молекулы (
- частоты
нормальных колебаний;
n - колебательные квантовые
числа,
h–
постоянная Планка, М
- приведенная масса ядра.
Нижний уровень энергии колебательного
движения молекулы (![]() =
0) называют нулевым колебательным
уровнем, а соответствующую ему энергию
- энергией нуле-вых колебаний молекулы.
=
0) называют нулевым колебательным
уровнем, а соответствующую ему энергию
- энергией нуле-вых колебаний молекулы.
Частоты колебаний, рассчитанные после неэмпирического решения уравнений Хартри-Фока с оптимизацией геометрии, согласуются с экспери-ментальными в пределах 3-10%. Для корректного расчета необходимо использовать базис 6-31G* и шире и учитывать корреляцию электронов. В ряде случаев хорошие результаты дают специально параметризованные полуэмпирические методы. Один из путей повышения точности расчетов - учет ангармонизма колебаний с использованием потенциала Морзе и других потенциалов.
Вращательная энергия. Квантовая механика в приближении жесткого ротатора дает для энергии вращательного движения линейной молекулы выражение [10, 11]
i = hBJ(J+1), (98)
где
![]() - вращательная постоянная; I=ΣmiRi2
- момент инерции молекулы, J =0, 1, 2…-
вращательное
квантовое число. Полученные неэмпирическим
расчетом равновесные длины связей Ri
позволяют вычислить составляющие
моментов инерции молекулы вдоль осей
симметрии (или вдоль некоторых выбранных
осей) и определить вращательную энергию
многоатомной молекулы.
- вращательная постоянная; I=ΣmiRi2
- момент инерции молекулы, J =0, 1, 2…-
вращательное
квантовое число. Полученные неэмпирическим
расчетом равновесные длины связей Ri
позволяют вычислить составляющие
моментов инерции молекулы вдоль осей
симметрии (или вдоль некоторых выбранных
осей) и определить вращательную энергию
многоатомной молекулы.
До сих пор речь шла о единственной молекуле. Абсолютное же большинство экспериментальных данных относится к свойствам, усредненным по всем состояниям (конфигурациям). Поэтому для корректного сравнения необходимо привлечение понятий статистической механики.
Рассмотрим вклады в молекулярную сумму по состояниям Q, принимая, что для рассматриваемых систем справедливо распределение Больцмана (классический предел). Тогда
Q = Qпост Qвращ Qкол Qэл . (99)
Поступательная сумма по состояниям Qпост определяется молекулярной массой m, температурой Т и объемом V (все термодинамические характеристики приводятся далее для одного моля вещества):
![]() .
(100)
.
(100)
Здесь k - постоянная Больцмана, m – масса молекулы.
Колебательная сумма по состояниям нелинейной n-атомной молекулы Qкол есть произведение
 .
(101)
.
(101)
Сумма по состояниям вращательного движения многоатомной жесткой молекулы с главными центральными моментами инерции IA , IB , IC при замене суммирования интегрированием равна [11]
 .
(102)
.
(102)
Электронную сумму по состояниям находят непосредственным суммированием
Qэл
=
![]() , (103)
, (103)
p0, p1, p2 - веса (степени вырождения) электронных уровней с учетом результирующего электронного спина молекулы (для многоатомных нелинейных молекул указанные веса согласно теореме Яна-Теллера определяется только мультиплетностью) [2].
Зная молекулярные суммы по состояниям, можно далее рассчитать вклады в термодинамические параметры в классическом пределе. Соответствующие соотношения приведены ниже:
Внутренняя
энергия: ![]() .
(104)
.
(104)
Энтальпия:
![]() .
(105)
.
(105)
Энергия
Гельмгольца: ![]() .
(106)
.
(106)
Энергия
Гиббса: ![]() .
(107)
.
(107)
Энтропия:
![]() .
(108)
.
(108)
Теплоемкость
изохорная:  .
(109)
.
(109)
Теплоемкость
изобарная:  .
(110)
.
(110)
Эти формулы записаны таким образом, чтобы явно указать необходимость выбора начального состояния. Нулевая энергия - средняя энергия системы при наименьших значениях квантовых чисел – является также энергией системы при Т = 0. К состоянию с нулевой энергией Е0 и следует отнести соответствующие значения внутренней энергии U0, энтальпии H0, энергии Гиббса G0 и Гельмгольца A0 данного индивидуального вещества, указывая вместе с тем и способ отсчета энергии:
U0 = A0 = E0 , (111)
H0 = G0 , (112)
H - H0 = (U - U0) + RT . (113)