

8. Влияние учета электронной корреляции на точность расчета. Расчет энергии диссоциации химических связей
Учетом кулоновской электронной корреляции и увеличением размера и гибкости базисного набора можно приблизится, в принципе, к точному ответу. Особенно это важно при исследовании эффектов, зависящих от возбужденных состояний или плохо описываемых в однодетерминантном приближении (пример - описание синглет-триплетного расщепления уровней метилена СН2 – табл. 6).
Таблица 6
Расчетные значения синглет-триплетного расщепления уровней метилена (ккал/моль), полученные разными методами в разных базисных наборах. (Эксп. значение 9.2 ккал/моль)
|
Метод/ базис |
6-31G |
6-31G* |
6-31G** |
6-311 G** |
6-311 ++G** |
6-311++ (3df,2pd) |
|
НХФ |
33.9 |
27.6 |
27.6 |
26.0 |
25.5 |
24.7 |
|
ОХФ |
36.8 |
30.9 |
30.9 |
29.3 |
28.8 |
28.1 |
|
MP2 |
28.7 |
20.8 |
20.1 |
18.0 |
17.4 |
15.0 |
|
MP3 |
26.4 |
18.3 |
17.5 |
15.3 |
14.8 |
12.6 |
|
MP4D |
25.3 |
17.1 |
16.3 |
14.1 |
13.5 |
11.3 |
|
MP4DQ |
25.4 |
17.3 |
16.6 |
14.4 |
13.9 |
11.8 |
|
MP4SDTQ |
25.2 |
17.0 |
16.2 |
14.1 |
13.5 |
11.4 |
Энергия диссоциации химической связи - энергия, необходимая для разрыва молекулы в том месте, где была связь. Ее расчет требует учета корреляции электронов: необходимо корректно описать весьма малые изменения в волновых функциях фрагментов, на которые диссоциировала молекула. Учет этих слабых возмущений эквивалентен учету электронной корреляции путем включения в расчет возбужденных электронных конфигураций.
Таблица 8
Энергии диссоциации (ккал/моль) некоторых молекул, рассчитанные
различными методами [3]
|
Реакция |
HF/6-31FG** //HF6-31G* |
MP2/6-31G** //MP2/6-31G* |
MP3/6-31G** //MP3/6-31G* |
MP4/6-31G** //MP4/6-31G* |
Эксп. |
|
H2H*+H* |
85 |
101 |
105 |
106 |
109 |
|
LiН Li*+H* |
32 |
45 |
48 |
49 |
58 |
|
BeH Be*+H* |
52 |
52 |
49 |
47 |
50-56 |
|
BH B*+H* |
62 |
77 |
79 |
80 |
82 |
9. Иерархия методов квантовой химии










Точный расчет
Учет
все большего числа возбужденных
электронных конфигураций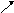
ССП
КВ методы, теория возмущений
Метод Хартри-Фока
ССП методы,
использующие НДП
(CNDO,
INDO, MINDO, и
др.
-электронное
приближение
Широкий
базис с учетом поляризационных и
диффузных функций
Метод
Хюккеля Минимальный
базис
Расширенный
базис
Полуэмпирические
методы
Рис. 8. Иерархия методов квантовой химии
Чтобы решить уравнения ХФ на практике используют приближение Борна-Оппенгеймера (задают структуру молекулы в виде координат ядер),
вводят приближение МО ЛКАО и выбирают аналитические функции, которыми будут аппроксимироваться АО. Эти функции называются базисными (или просто базисом). Далее проводится строгий самосогласованный расчет с вычислением всех необходимых интегралов. Такой способ вычисления МО называется неэмпирическим или ab initio, т.е. из первых принципов.
Когда исследуются ряды соединений и важны лишь относительные значения энергии и других характеристик, можно оценивать интегралы в схеме расчета на основании экспериментальной информации. Такие методы называются полуэмпирическими: они значительно проще и быстрее неэмпирических методов, а подчас дают и лучшие результаты. Это достигается за счет удачной параметризации и одновременно определяет основной недостаток полуэмпирических методов - плохую переносимость параметров от одного класса соединений к другому.