

Multiple Bonds Between Metal Atoms / 12-Rhodium Compounds
.pdfRhodium Compounds 555
Chifotides and Dunbar
12.69
A cage complex [{NEt4 [cis-Rh2(DAniF)2L]4[calix[4]arene(CO2)4]2}]BF4 (Fig. 12.45), that is capable of permanently encapsulating imprisoned guest molecules, is formed by capping four [Rh2(cis-DAniF)2]22+ corner piece precursors with two anions of the toroidal or chalicelike calix[4]arenetetracarboxylate ligands through the eight carboxylate groups of the two ligands.484 The dirhodium units serve as ‘fasteners’ that hold together the two bowls of the calixarenes. The resulting carceplex encapsulates tetraethylammonium ions rather selectively to afford a stable species that remains intact in solution, even under the conditions necessary for mass spectroscopy.
Fig. 12.45. Schematic drawing showing the formation of the carceplex [{NEt4 [cis- Rh2(DAniF)2L]4[calix[4]arene(CO2)4]2}]BF4 (left) and the molecular structure (right). The NEt4 ion is encapsulated in the cavity.
12.7.3 Biological applications of dirhodium compounds
The extraordinary success of cis-Pt(NH3)2Cl2 (cisplatin) as a leading metal-based antitumor drug,749-751 ushered to a new era in the development of other chemotherapeutic anticancer agents752 with improved specificity, reduced toxicity and cell resistance. Among the promising non-platinum antitumor complexes are dirhodium compounds, a fact that has spawned a number of investigations of their biological effects upon encountering plausible cellular targets such as DNA, polymerases or other proteins.753,754 Although their precise antitumor mechanism of action has not been elucidated, it has been demonstrated that dirhodium compounds, in a manner akin to that of cisplatin, bind to DNA755-759 and inhibit DNA and protein synthesis.177,760-764
Pioneering studies that emanated in the 1970’s showed that dirhodium carboxylate compounds Rh2(O2CR)4 (R = Me, Et, Pr) exhibit significant in vivo antitumor activity against L1210 tumors,765,766 Ehrlich ascites,755,756,767-769 sarcoma 180 and P388 tumor lines.770 It is notable that the antitumor activity increases in the series Rh2(O2CR)4 (R = Me, Et, Pr) with the lipophilicity of the R group, but further lengthening of the carboxylate moiety beyond the pentanoate reduces the drugs’ therapeutic efficacy;756,766,767,771 this increase is independent of their

556Multiple Bonds Between Metal Atoms Chapter 12
redox properties.772 A small increase in effectiveness is achieved by modifications such as using the polyadenylic acid adduct of Rh2(O2CC2H5)4 or [Rh2(O2CC2H5)4]+ instead of Rh2(O2CC2H5)4 itself.772,773 Moreover, dirhodium carboxylate compounds and their nitroimidazole adducts have been reported to increase the radiation sensitivity of hypoxic mammalian774,775 and bacterial776-778 cells in vitro.
A systematic variation of the ax and eq ligands on the dirhodium unit has provided valuable insight into the structure-activity relationships for this family of compounds. Substitution of the carboxylate (pKb = 9.25) bridging groups of Rh2(O2CCH3)4 with trifluoroacetate (pKb > 13) renders the dirhodium compound more reactive due to the greater lability and the strong electron withdrawing effect of the CF3CO2- groups. It has been reported that Rh2(O2CCF3)4 and its adduct with sulfadiazine significantly increase the survival rate of mice bearing Ehrlich ascites cells and induce a higher mortality for these tumor cells in vitro.779 When the basic trifluoroacetamidate bridging group CF3CONH (12.5) is introduced into the dirhodium core, a 90% survival rate of the Ehrlich tumor-bearing population and an LD50 value of the same order as that of cisplatin to promote the same inhibitory effects on cell growth, are observed.780,781 Cationic compounds of general formulae [Rh2(µ-O2CCH3)2(N−N)2(H2O)2]2+ (N−N = 2,2' bipyridine (bpy) or 1,10 phenanthroline (phen)) exhibit anticancer activity against human oral carcinoma KB cell lines comparable to Rh2(O2CCH3)4782 and, appreciable antibacterial activity.783-786 Dirhodium compounds with tridentate oxygen-metalated methoxyphenylphosphine groups exhibit improved antitumor activity compared to dirhodium tetraacetate; the most active member of the series, Rh2(µ-O2CCH3)3[µ-(o-OC6H4)P(o-OMeC6H4)2](HOCCH3),568 exhibits higher antitumor activity than cisplatin against several cell lines.543 The compound cis-Rh2(DTolF)2(O2CCF3)2(H2O)2 with two robust formamidinate (12.7) and two labile trifluoroacetate bridging groups represents a favorable compromise between antitumor activity and toxic side-effects; it exhibits comparable antitumor activity to that of cisplatin against Yoshida ascites and T8 sarcomas with considerably reduced toxicity.443 The homoleptic paddlewheel compound Rh2(DTolF)4, however, exhibits no appreciable biological activity,787 presumably due to steric factors which preclude access of biological targets to ax and eq sites of the dirhodium core. Alternative strategies for improving dirhodium drug activity have paved the way to designing complexes with water soluble ligands such as carbohydrate and cyclophosphamide derivatives,788,789 compounds with the dirhodium core attached to carrier ligands such as isonicotinic acid,790,791 the substituted triazene Berenil,169 metronidazole,177 organic antimalarial drugs,792 and cyclodextrin encapsulated compounds;793,794 the latter offer a useful method for localized and controlled release of the drug with minimized side-effects.795
Human serum albumin (HSA) has been proposed as a pertinent transport protein for dirhodium carboxylate compounds796,797 since it has been found to readily form adducts with the latter, most likely via the imidazole rings of the histidine residues.798,799 The compound cis-[Rh2(µ-O2CCH3)2(bpy)2(H2O)2](O2CCH3)2 also interacts with HSA and causes alterations to the secondary structure of the protein,800 similarly to tetracarboxylate compounds.796,798 The interactions of dirhodium compounds vis-à-vis several enzymes and sulfur-containing biomolecules have been explored owing to their biological relevance, but the conclusions have not been unequivocally established.252,253,760,801-805
Interactions with nucleobases, nucleos(t)ides and DNA
Nucleobases and nucleosides.6 The reactions of dirhodium compounds with purine nucleobases (12.70 and 12.71) and nucleos(t)ides have received considerable attention because DNA is the primary target of most metal-based anticancer agents.806 A perusal of the literature reveals that dirhodium compounds exhibit a strong preference for binding to adenine (12.70) compared

Rhodium Compounds 557
Chifotides and Dunbar
to guanine (12.71).181,182,184,186,187,190-192,755,756,807-810 The immediate color change from green (or blue) to violet (or pink) upon replacing ax O donors with N donors attests to the interaction of dirhodium compounds with adenine and its derivatives. Binding of adenine bases predominantly takes place via N7 and N1 (12.70), which typically results in formation of polymeric bridged compounds of type a (Fig. 12.2);182,184,190,807 in the cases of steric hindrance182 or substitution of N1,184 binding takes place via N7 leading to adducts of type 12.11.
The crystal structure determinations of Rh2(O2CCH3)4(1-MeAdo)2184 and trans-[Rh2(µ- O2CCH3)2(µ-HNCOCF3)2(9-MeAdeH2)2](NO3)2411 indicate that preferential binding of adenine via N7 may be attributed to intramolecular hydrogen bonds established between the exocyclic NH2(6) amino group of the purine and the carboxylate oxygen atoms (12.73).190,807 The argument is further supported by the fact that the guanine analog theophylline (12.72) binds axially to Rh2(O2CCH3)4 via N9 to avoid electrostatic repulsion between the O6 exocyclic carbonyl group and the carboxylate oxygen atoms.185 Conversely, in the case of [Rh2(HNCOCH3)4(theophylline)2]NO3,715 theophylline binds via N7 due to the favorable hydrogen bonding interactions between the theophylline site O6 and the NH hydrogen-donor group of the bridging acetamidate ligand. Similar favorable interactions have been established for dirhodium-acetamidate ax adducts of cytosine,410 guanine (12.71) and its nucleosides,411 as well as for the dirhodium adduct with the biologically relevant drug azathioprine.188
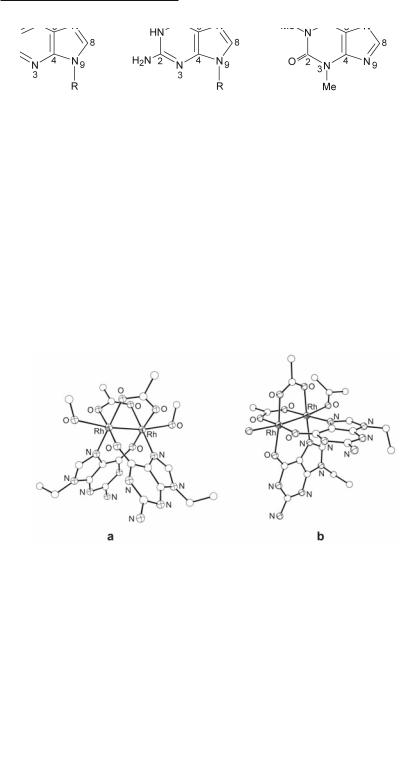
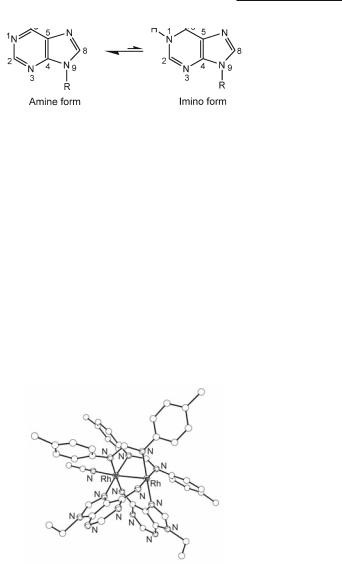
Electrostatic repulsion between the carboxylate oxygen atoms and the O6 exocyclic carbonyl group of guanine is responsible for its reduced reactivity towards Rh2(O2CCH3)4, a conclusion that is supported by the lack of perceptible color change upon Rh2(O2CCH3)4 reaction with guanine and its derivatives. This behavior led to early claims in the literature that dirhodium carboxylate compounds do not react with guanine and polyguanylic acids.755,807 The issue was settled, however, with the crystal structural determinations of H-T cis-[Rh2(µ-O2CCH3)2(9- EtGua)2(MeOH)2],393 (Fig. 12.46a), H-H cis-[Rh2(µ-O2CCH3)2(9-EtGuaH)2(Me2CO)(H2O)]- (BF4)2394 (Fig. 12.46b) and H-T cis-[Rh2(µ-O2CCF3)2(9-EtGuaH)2(Me2CO)2](CF3CO2)2,393 which revealed unprecedented bridging guanine groups that span the dirhodium unit via the N7/O6 sites in a cis disposition and H-H (12.28) or H-T (12.29) orientations. These crystal structures provided the first hard evidence for guanine O6 participation in binding to dimetal units. Notable features of H-T cis-[Rh2(µ-O2CCH3)2(9-EtGua)2(MeOH)2] are the deprotonation of the purine site N1, i.e., the enolate form of guanine (9-EtGua–) is stabilized393 (12.74-12.75) and the substantial increase in the acidity of N1–H due to bidentate N7/O6 coordination (pH dependent 1H and 13C NMR titrations afford a pKa value of c. 5.7 compared to 8.5 for N7-bound only and 9.5 for the unbound purine).811 The importance of the O6/N1 guanine sites is obvious, given that they are involved in Watson-Crick hydrogen bonding in duplex DNA; alteration of these sites would lead to DNA base mispairing which bears directly on metal mutagenicity and cell death.812 Bridging 9-EtGuaH and 9-EtAdeH groups, spanning the dirhodium unit in a similar fashion via N7/O6 (12.71) and N7/N6 (12.70), respectively, have been observed in H-H cis-[Rh2(DTolF)2(9-EtGuaH)2(NCCH3)](BF4)2462 and H-T cis-[Rh2(DTolF)2(9-EtAdeH)2(NCCH3)](BF4)2461,462 (Fig. 12.47) wherein the dirhodium unit is supported by DTolF groups. In the case of H-T cis-[Rh2(DTolF)2(9-EtAdeH)2(NCCH3)](BF4)2, 9-EtAdeH (12.76) is present in the rare imino form 12.77, as suggested by variable temperature 1H NMR spectra.461 The presence of the adenine imino form in DNA may lead to alteration of the base hydrogen bonding behavior and an increase in the acidity of N1-H, ultimately causing nucleobase mispairing and cell mutations.812,813
558Multiple Bonds Between Metal Atoms Chapter 12
12.70 |
12.71 |
12.72 |
12.73
Fig. 12.46. The structures of (a) H-T cis-[Rh2(µ-O2CCH3)2(9-EtGua)2(MeOH)2] and
(b) the cation in H-H cis-[Rh2(µ-O2CCH3)2(9-EtGuaH)2(Me2CO)(H2O)](BF4)2.
12.74 |
12.75 |
Rhodium Compounds 559
Chifotides and Dunbar
12.76 |
12.77 |
The aforementioned crystal structures argue strongly for the prevalence of this eq bridging binding mode of guanine and adenine bases with dinuclear compounds. The crystal structure determination of cis-[Rh2(µ-O2CCH3)2(bpy)(9-EtGuaH)(H2O)2(CH3SO4)]CH3SO4 395 revealed, however, that 9-EtGuaH may also bind in a monodentate fashion via N7 to a single rhodium center at an eq position, in the presence of a chelating agent (bpy) which occupies eq sites of the other rhodium center.393,394 These findings provide insight into the possible mechanism of interaction between dirhodium compounds and biologically relevant nucleobases or nucleos(t)ides. This chemistry most likely involves initial attack of the nucleophilic base at the ax position of the dimetal core to afford an axially bound monodentate adduct followed by rearrangement to eq sites, as has been observed in the case of chelating N-N donor ligands (e.g., bpy; Fig. 12.17).373 The rearrangement of ligands from ax to eq positions is a key feature in dictating the outcome of purine reactions with dirhodium units.
Fig. 12.47. Structure of the cation in H-H cis-[Rh2(DTolF)2(9-EtAdeH)2(NCCH3)](BF4)2.
A natural extension of dirhodium reactions with model nucleobases is that with small DNA fragments. Early studies report the stepwise formation constants of 1:1 and 1:2 adducts of Rh2(O2CCH3)4 with adenine nucleotides, but the compounds were not isolated.181 Subsequent studies based on 1H NMR and infrared spectroscopies are consistent with ax binding of adenine nucleotides via N7 and N1 to the dirhodium core.182,814 Studies on the reaction of Rh2(O2CCH3)4 with guanosine-5'-monophosphate (GMP; 12.78 for X = PO3H-), performed by 1H and 13C NMR spectroscopies, suggest the formation of two isomers with the guanine rings spanning the Rh–Rh bond in a bridging fashion via N7/O6 and H-T or H-H arrangement of the bases, as in the case of 9-EtGuaH (Fig. 12.46a and 12.46b, respectively).815
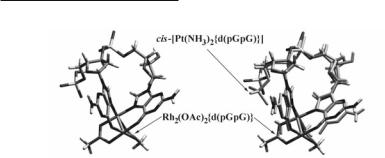
Extension of the knowledge obtained from the dirhodium unit interactions with the basic building blocks of DNA, led to the reasonable hypothesis that the 90° ‘bite’ angle displayed by the d(GpG)-cisplatin ‘chelate’ is well suited to accommodate two cis eq positions of one metal

560Multiple Bonds Between Metal Atoms Chapter 12
atom in a dirhodium unit, despite the different geometries of the two metal complexes. Indeed, reactions of Rh2(O2CCH3)4 with the dinucleotides d(GpG) (12.79; X = H) and d(pGpG) (12.79; X = PO3H-) afford Rh2(O2CCH3)2{d(GpG)} (12.80; X = H) and Rh2(O2CCH3)2{d(pGpG)} (12.80; X = PO3H-), respectively, with bidentate N7/O6 bridging bases spanning the Rh–Rh bond.811,815 For both dinucleotide complexes, intense H8/H8 ROE (Rotating frame nuclear Overhauser Effect) cross-peaks in the 2D ROESY NMR spectrum (Fig. 12.48) indicate H-H arrangement of the guanine bases (12.80).811,815 The Rh2(O2CCH3)2{d(GpG)} complex exhibits two major right handed conformers HH1R (~ 75%) and HH2R (~ 25%), which differ in the relative canting of the two bases.811 In the case of Rh2(O2CCH3)2{d(pGpG)}, the presence of the terminal 5'-phosphate group results in stabilization of only one left-handed Rh2(O2CCH3)2{d(pGpG)} HH1L conformer due to the steric effect of the 5'-group favoring left canting, as in cisplatin-DNA adducts.815 Detailed characterization of Rh2(O2CCH3)2{d(GpG)}811 and Rh2(O2CCH3)2{d(pGpG)}815 by 2D NMR spectroscopy, revealed notable structural features that resemble those of cis-[Pt(NH3)2{d(pGpG)}]; the latter involve repuckering of the 5'-G sugar rings to the C3'-endo (N-type) conformation, retention of the C2'-endo (S-type) conformation for the 3'-G sugar rings and anti orientation of the bases with respect to the glycosyl bonds. The superposition of the low energy Rh2(O2CCH3)2{d(pGpG)} conformer (Fig. 12.49a), generated by simulated annealing calculations, and the crystal structure of cis- [Pt(NH3)2{d(pGpG)}]816 reveals remarkable similarities between the adducts (Fig. 12.49b); not only are the bases almost completely destacked (interbase dihedral angle 3'-G/5'-G 5 80°) upon coordination to the metal in both cases, but they are favorably poised to accommodate the bidentate N7/O6 binding to the dirhodium unit.815 Contrary to conventional wisdom, two metal-metal bonded rhodium atoms are capable of engaging in cis binding to GG intrastrand sites by establishing N7/O6 bridges that span the Rh–Rh bond. The rigid steric demands of the tethered guanine bases bound to the square planar platinum atom in cis-[Pt(NH3)2{d(pGpG)}] are also satisfied in metal-metal bonded dirhodium units. Our unprecedented findings that d(GpG) fragments establish bridging eq bonds via N7/O6 with the dirhodium core reveal new possibilities for metal-DNA interactions and lay a solid foundation for exploring similar structural motifs in related systems. Indeed, 1D and 2D NMR spectroscopic data of the dirhodium formamidinate dinucleotide complexes Rh2(DTolF)2{d(GpG)}, Rh2(DTolF)2{d(ApA)}, Rh2(DTolF)2{d(GpA)} and Rh2(DTolF)2{d(ApG)} (d(XpX) involves two purine bases X linked with a phosphodiester bond, X = adenine, 12.70; guanine, 12.71), corroborate N7/O6 and N7/N6 binding of the guanine and adenine rings, respectively, with H-H arrangement of the tethered nucleobases.817 Contrary to cis-[Pt(NH3)2{d(pGpG)}] and Rh2(O2CCH3)2{d((p)GpG)}, in the case of Rh2(DTolF)2{d(GpG)} both sugar rings are of type N, a fact that implies possible conformational restriction for the compound. Variable temperature 1H NMR studies of Rh2(DTolF)2{d(ApA)} indicate that the adenine bases are present in the rare imino form 12.77, as in the case of H-T cis-[Rh2(DTolF)2(9-EtAdeH)2(NCCH3)](BF4)2.

Rhodium Compounds 561
Chifotides and Dunbar
12.80
Fig. 12.48. Aromatic region of the 2D ROESY NMR spectrum of Rh2(O2CCH3)2{d(pGpG)}, in D2O at 5 °C, pH 7.8, displaying the H8/H8 ROE crosspeaks of the two guanine bases in a H-H arrangement.
562Multiple Bonds Between Metal Atoms Chapter 12
a |
b |
Fig. 12.49. (a) Low energy Rh2(O2CCH3)2{d(pGpG)} conformer resulting from simulated annealing calculations; (b) Superposition of the crystallographically determined cis-[Pt(NH3)2{d(pGpG)}] and the lowest energy Rh2(O2CCH3)2{d(pGpG)} conformer.
Oligonucleotides. Reactions of Rh2(O2CCH3)4, [Rh2(O2CCH3)2(NCCH3)6]2+, and Rh2(O2CCF3)4 with single-stranded oligonucleotide tetramers, octamers and dodecamers containing AA, GG, GA and GA dipurine sites, e.g., d(TGGT), d(TTCAACTC), d(CCTCTGGTCTCC), point to the following relative order of reactivity associated with the lability of the leaving groups: cis- [Pt(NH3)2(H2O)2]2+ (activated cisplatin) ~ Rh2(O2CCF3)4 > cis-[Pt(NH3)2Cl2] (cisplatin) >> cis- [Rh2(O2CCH3)2(NCCH3)6](BF4)2 > Rh2(O2CCH3)4.758 Bis-acetate oligonucleotide adducts are the dominant species for the tetramers, whereas for longer oligonucleotides, the monoacetate and dirhodium species with no acetate bridging groups are detected. Although the metabolism of dirhodium carboxylate compounds in mammals involves displacement of one or more acetate bridges which are eventually oxidized to CO2,818 the dirhodium species detected in the aforementioned mass spectrometry study along with the kinetic stabilities of cis-[Rh2(NCCH3)10]4+ and [Rh2(H2O)10]4+ 596 suggest that the dirhodium core remains intact in the absence of carboxylate bridging groups. Electrospray ionization mass spectrometry permitted the observation of initial ax dirhodium-DNA adducts, followed by rearrangement to stronger eq-DNA species.758 The detection of these adducts provides insight into the mechanism of interaction of dirhodium units with short oligonuleotides; the latter is most likely similar to that observed in the case of chelating N-N donor ligands, e.g., bpy; Fig. 12.17.373 Enzymatic digestion studies with 3'Α5' DNA and 5'Α3' DNA exonucleases (Phosphodiesterase I and II, respectively) followed by MALDI and ESI MS, indicate that dipurine rather than pyrimidine sites of DNA oligonucleotides preferentially bind to the dirhodium core.757,758
Double-stranded (ds) DNA. Polyacrylamide Gel Electrophoresis (PAGE) studies address the longstanding issue of if and how dirhodium compounds bind to dsDNA. These studies refute earlier claims that no reaction between dirhodium compounds and dsDNA occurs, and indicate that interaction of dsDNA with dirhodium carboxylate compounds leads to covalent cross-linking of the two DNA strands.819 The extent of DNA interstrand cross-link formation correlates with the lability of the leaving groups and is in the order Rh2(O2CCF3)4 > [Rh2(O2CCH3)2- (NCCH3)6](BF4)2 >> Rh2(O2CCH3)4. The reversal behavior of the dsDNA interstrand cross-links in 5 M urea at 95 °C implies the presence of a mixture of monofunctional and/or bifunctional ax/ax, ax/eq or eq/eq dirhodium-DNA adducts. The less stable adducts in the isolated band are most likely ax-DNA adducts which are expected to exhibit enhanced exchange rates with heating compared to eq-DNA species.596 The reversal of additional dsDNA-dirhodium adducts in the isolated band, by further heating in 40 mM thiourea, indicates the presence of another subset of products that are stable to more harsh conditions (most likely eq-DNA adducts).819 These studies provide valuable insight into the possible underlying mechanism(s) of dirho-

Rhodium Compounds 563
Chifotides and Dunbar
dium antitumor behavior. In conclusion, it is important that DNA be considered a potential biological target of dirhodium compounds; the data obtained thus far constitute an excellent backdrop for further biochemical studies of metal-metal bonded systems.
Photochemistry and DNA photocleavage
Various dirhodium complexes have been investigated as potential antitumor agents in photochemotherapy, which involves triggering the toxicity of a compound by irradiation of the affected area with low energy visible or near-ir light to permit better tissue penetration.
It has been reported that Rh2(O2CCH3)4 exhibits a long-lived excited state (T = 3.5 µs) that can be accessed with visible light (ηexc ~ 350–600 nm) and is able to undergo energy and electron transfer with a variety of acceptors.7,8 Irradiation of Rh2(O2CCH3)4 with visible light (ηirr = 400–610 nm), in the presence of electron acceptors, results in DNA photocleavage by the mixed-valent cation [Rh2(O2CCH3)4]+ (Fig. 12.50).9,820 The absorption of the related formamidinate (12.7) compounds Rh2(ArNCHNAr)4, Ar = XC6H4 (X = p-OMe, p-CF3, p-Cl), as well as that of Rh2(tpg)4 (Fig. 12.26) extends to c. 880 nm, and irradiation of these complexes in the presence of various alkyl halide substrates results in formation of the corresponding mixed-valent Rh25+ compounds Rh2(ArNCHNAr)4X (X = Cl, Br);500,821 the compound Rh2(DAniF)4Cl has been structurally characterized (Table 12.9).500 The aforementioned complexes also effect DNA photocleavage in the presence of electron acceptors.500
Fig. 12.50. Imaged agarose gel showing the photocleavage of 100 µM pUC18 plasmid (5 mM Tris buffer, pH 7.5) by 40 µM Rh2(O2CCH3)4(H2O)2 (Rh2) in the presence of 2 mM py+,10 min irradiation, ηirr 395 nm. Lane 1: plasmid only, dark. Lane 2: plasmid only, irradiated; Lane 3: plasmid + Rh2, irradiated; Lane 4: plasmid +py+, irradiated; Lane 5: plasmid + Rh2 + py+, dark; Lane 6: plasmid + Rh2 + py+, irradiated; 3-cyano-1-methyl- pyridinium tetrafluoroborate: py+ (electron acceptor).
Unlike Rh2(O2CCH3)4, which requires an electron acceptor in solution, Rh24+ complexes with dppz (dppz: dipyrido[3,2-a:2',3'-c]phenazine), cis-[Rh2(µ-O2CCH3)2(dppz)(δ1- O2CCH3)(CH3OH)]+(12.81)andthestructurallycharacterized822cis-[Rh2(µ-O2CCH3)2(dppz)2]2+
(12.82), photocleave pUC18 plasmid in vitro upon near-uv (ηirr 320 nm) and visible (ηirr 395 nm) irradiation, resulting in the nicked, circular DNA (Fig. 12.51; lanes 4 and 6, respectively). An enhanced degree of photocleavage is observed for the former compared to the latter, which may be due to the ability of cis-[Rh2(µ-O2CCH3)2(dppz)(δ1-O2CCH3)(CH3OH)]+ to intercalate DNA bases.823 The compounds cis-[Rh2(µ-O2CCH3)2(dppn)2]2+ 824 (dppn: benzo[i]dipyrido[3,2- a:2',3'-c]phenazine) and cis-[Rh2(µ-O2CCH3)2(dppz)2]2+ 822 (Table 12.2) exhibit relatively low cytotoxicities in the dark, but their toxicities increase significantly (by 24and 3.4-fold, re-

564Multiple Bonds Between Metal Atoms Chapter 12
spectively) when the cell cultures are irradiated with visible light (400-700 nm, 30 min); the cation cis-[Rh2(µ-O2CCH3)2(dppn)2]2+ has the added advantage of 18-fold lower toxicity than hematoporphyrin (key component in Photofrin©) in the dark. Likewise, the cytotoxicity of the heteroleptic species cis-[Rh2(µ-O2CCH3)2(bpy)(dppz)]2+ (Fig. 12.52) increases 5-fold upon irradiation, with the advantage of 10and 7.5-fold lower toxicity than hematoporphyrin and cis-[Rh2(µ-O2CCH3)2(dppz)(δ1-O2CCH3)(CH3OH)]+, respectively, in the dark.825 The substitution of two eq labile groups in cis-[Rh2(µ-O2CCH3)2(dppz)(δ1-O2CCH3)(CH3OH)]+ with a chelating bpy moiety apparently leads to reduction of its toxicity. The latter results render these compounds promising candidates for photochemotherapy. In the dirhodium series cis-[Rh2(µ-O2CCH3)2(dppz-X2)2]2+ (X = OCH3, CH3, Cl, NO2) with substituted dppz (at positions 7 and 8 of the dppz ring), the percent of effected DNA photocleavage increases as the electron-donating ability of the group X increases.826 The formamidinate derivatives cis-[Rh2(DPhFF)2(dppz)(NCCH3)4]2+ (Fig. 12.53) and cis-[Rh2(DPhFF)2(dppz)2(NCCH3)2]2+ have also been prepared and will be the subject of future studies.827 The ultimate goal of these investigations is to effectively control the photoreactivity and cytotoxicity of the previous compounds by tailoring both the eq bridging groups as well as the other ligands on the dimetal unit. Preliminary studies indicate that the cytotoxicity of dirhodium carboxylate compounds towards healthy human skin cells increases by substitution of acetate with the more labile trifluoroacetate bridging groups.828
12.81 |
12.82 |
Fig. 12.51. Imaged agarose gel (2%) exhibiting the photocleavage (ηirr 395 nm, 20 min) of 100 µM pUC18 plasmid. Lane 1: plasmid only, dark; Lane 2: plasmid treated with Smal to produce linear DNA; Lane 3: plasmid treated with: 10 µM cis- [Rh2(µ-O2CCH3)2(dppz)(δ1-O2CCH3)(CH3OH)]+, dark ; Lane 4: plasmid treated with 10 µM cis-[Rh2(µ-O2CCH3)2(dppz)(δ1-O2CCH3)(CH3OH)]+, irradiated; Lane 5: plasmid treated with 10 µM cis-[Rh2(µ-O2CCH3)2(dppz)2]2+ , dark; Lane 6: plasmid treated with10 µM cis-[Rh2(µ-O2CCH3)2(dppz)2]2+, irradiated.