

Multiple Bonds Between Metal Atoms / 12-Rhodium Compounds
.pdfRhodium Compounds 525
Chifotides and Dunbar
compounds. Such (N, P) bridging groups spanning the dirhodium unit are encountered in Rh2[µ-PhP(py2)]2Cl4.516 Electrochemical oxidation of Rh2(µ-CO)Cl2(µ-Ph2Ppy)2 affords the unusual compound trans-Rh2(µ-Ph2Ppy)2(µ-NO3)(CO)Cl3 with two Ph2Ppy groups in H-T arrangement and a bridging nitrate ion.517 Electrochemical oxidation of trans-Rh2(µ-CO)Cl2(µ- Ph2Ppy)2, in the presence of chloride ions affords trans-Rh2(CO)Cl4(µ-Ph2Ppy)2, which is the starting material for trans-Rh2(CO)2Cl4(µ-Ph2Ppy)2.518 The Rh–Rh distance in trans-Rh2(µ- CO)Cl2(µ-Ph2Ppy)2 (2.612(1) Å)519 is essentially identical to that in the unusual heterobridged complex Rh2I2(CO)2(µ-pz)2(µ-dppm) (2.612(3) Å), which contains a Rh–Rh bond supported by one dppm group and two bidentate (N, N) donor groups.520 The Rh–Rh bond distance (2.725(2) Å) in trans-[Rh2I2(CO)2(3,5-Me2pz)(µ-dppm)2]ClO4 (12.36), which is prepared by oxidation of trans-[Rh2(CO)2(3,5-Me2pz)(µ-dppm)2]ClO4 with I2, is in the range of other similar A-frame compounds.521
Fig. 12.27. The core of Rh2(µ-Cl)2(µ-dppm)2Cl2.
12.32 |
12.33 |
12.34 |
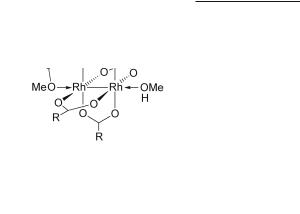
Reaction of Rh2(O2CCH3)4 with PPh3 in refluxing acetic acid leads to isolation of the orthometalated product Rh2(µ-O2CCH3)2[Ph2P(C6H4)]2(HO2CCH3)2 with two bridging acetate ligands in a cisoid arrangement and two bridging [Ph2P(C6H4)]< anions spanning the Rh–Rh bond in H-T orientation (12.37).522,523 The reaction proceeds through formation of Rh2(µ-O2CCH3)3[Ph2P(C6H4)](HO2CCH3)2 which reacts with an additional equiv of PPh3, to afford the doubly orthometalated product.524,525 Detailed studies that shed light into the role of the acid and the mechanism of the orthometalation reactions have been reported.526-529 A number of dirhodium complexes of general formulae Rh2(µ-O2CR)3(PC),374,525,530-537 and cis-Rh2(µ-O2CR)2(PC)2522,523,536-553 (PC stands for orthometalated arylphosphines) with both H-H and H-T arrangements have been reported554 to be highly active catalysts for certain intramolecular carbene insertion reactions (see Chapter 13). A structural characteristic of bisorthometalated complexes is the elongation of the Rh–Rh bond compared to Rh2(O2CR)4, the bond lengths ranging from 2.48 to 2.63 Å (Table 12.2). The longest Rh–Rh bonds are encountered in complexes with strong ax ligands such as PPh3 (2.630(1) Å)547 and the shortest in H-T cis-Rh2(µ-O2CCH3)2[PhP(C6H4)(o-BrC6F4)]2 (2.475(1) Å) wherein the bromine atom of
526Multiple Bonds Between Metal Atoms Chapter 12
the C6F4Br ring occupies one of the dirhodium core ax sites.555 Related compounds with chiral phosphines556 and phosphanes557-560 have been reported, as well as two dirhodium catalysts with two H-T metalated phosphine and two succinimidate (N, O donor) bridging groups.561
12.35 |
12.36 |
12.37 |
Reaction of Rh2(µ-O2CCH3)2[Ph2P(C6H4)]2 with Me3SiCl in warm THF and the monophosphine ligands PMe3 or PPh3 affords Rh2[Ph2P(C6H4)]2(µ-Cl)2(PMe3)2 or Rh2[Ph2P(C6H4)]2- (µ-Cl)2(PPh3)2, respectively, which retain the H-T cisoid arrangement of the starting material.562 A similar reaction of Rh2(µ-O2CCH3)2[Ph2P(C6H4)]2 in warm THF, in the presence of Me3SiCl and the bridging phosphine dmpm, affords Rh2(dmpm)2[Ph2P(C6H4)]2Cl2 which has two ax Cl− anions, two cis orthometalated bridging [Ph2P(C6H4)]− anions in H-T arrangement and two cis bridging dmpm groups that also span the Rh–Rh bond.507,563 The different outcome of the two previous reactions is attributed to the structure of dmpm, which precludes the chloride ligands from occupying bridging positions.563 The long Rh–Rh bond distance (2.770(3) Å) in Rh2(dmpm)2[Ph2P(C6H4)]2Cl2507,563 (Table 12.5), compared to tetracarboxylate (Table 12.1) and mixed carboxylate/orthometalated phosphine (Table 12.2) compounds, is attributed to loss of the small ‘bite’ carboxylate groups which act to hold the dirhodium unit together along with the addition of the ax chloride ligands.507
Reaction of cis-Rh2(µ-O2CCH3)2[Ph2P(C6H4)]2(HO2CCH3)2 with excess Me3OBF4 in CH3CN results in formation of racemic cis-{Rh2[Ph2P(C6H4)]2(NCCH3)6}(BF4)2 with two cisoid non-labile orthometalated phosphine bridging anions and six labile CH3CN ligands occupying eq and ax positions.564 This is the first structurally characterized orthometalated dirhodium compound in which the four additional eq positions (apart from those occupied by the bridging phosphine groups) are occupied by non-bridging ligands and, the first molecule with an inherently chiral metal-metal bonded unit.564 Reaction of cis-{Rh2[Ph2P(C6H4)]2(NCCH3)6}(BF4)2 with salts of linear dicarboxylate anions (e.g., oxalate, terephthalate) affords molecular triangles that employ orthometalated phosphine units as building blocks;564 these are discussed in Section 12.7.2.
The highly basic and bulky phosphine ligand 2,4,6-trimethoxyphenylphosphine (TMPP) reacts with dirhodium carboxylate compounds to afford unusual products due to the ‘noninnocence’ of the ether functionalities. Reaction of TMPP with Rh2(O2CCH3)4(MeOH)2 in the presence of ethanol, yields Rh2(µ-O2CCH3)3(O-TMPP)(MeOH) (12.38), wherein the dirhodium unit is spanned by a tridentate oxygen-metalated O-TMPP ligand (demethylation of a methoxy substituent on one of the phenyl rings of TMPP takes place).565,566 In the case of the TMPP reaction with the strong Lewis acid Rh2(O2CCF3)4, the ligand arrangement is similar to that in [Rh2(µ-O2CCH3)3(O-TMPP)], with one important difference; namely, the phenoxide oxygen involved in the six-membered ring Rh–Rh–P–C–C–O is also bonded to another unit of [Rh2(µ-O2CCF3)3(O-TMPP)] to afford the ‘dimer of dimers’ [Rh2(µ-O2CCF3)3(TMPP-O)]2.567 A tridentate oxygen-metalated binding mode, similar to that of TMPP in 12.38, is observed with 2-methoxyphenylphosphine (MPP) in Rh2(µ-O2CCH3)3(O-MPP)(HO2CCH3)516 and Rh2(µ- O2CCH3)3(O-MPP)(NCCH3).568 Under refluxing conditions, Rh2(µ-O2CCF3)2(TMPP-O)2 is
Rhodium Compounds 527
Chifotides and Dunbar
obtained with each O-TMPP bound to a Rh center in a face-capping mode through the phosphorus and ether oxygen atoms as well as a phenoxide group.567
12.38
The homologous series of Rh20, Rh22+, Rh24+ complexes with bridging bis(difluorophosphin o)methylamine and its trifluoroethoxy 569 derivatives have been prepared and structurally characterized using the compounds [RhCl(PF3)2]2,570-572 [RhBr(PF3)2]2,571,572 or [RhBr(C8H8)]211 as starting materials. Their electronic absorption spectra are dominated by intense bands that are characteristic of dμ Α dμ* transitions and each compound exhibits an emissive dμ* excited state.570,571 The increase of the Rh–Rh and Rh–X distances in the Rh2[CH3N(PF2)2]3X4 family of compounds, upon replacement of Cl with Br, is consistent with the increasing size of the bridging atom.572,573
12.3.6 Complexes supported by carbonate, sulfate and phosphate bridging groups
Dirhodium compounds with bridging HCO3−, CO32−, H2PO4−, HPO42−, HSO4−, and SO42− anions have all been reported. Among them, carbonate compounds are similar to carboxyl- ate-bridged compounds and will be discussed first. The structurally characterized dirhodium compounds with the aforementioned bridging groups are compiled in Table 12.6.
Table 12.6. Dirhodium compounds with bridging carbonate, sulfate and phosphate groups
Compound |
r (Rh–Rh)a (Å) |
r (Rh–Lax) (Å) |
Donor |
ref. |
|
atom(s) |
|||||
|
|
|
|
||
Cs4[Rh2(CO3)4(H2O)2]·6H2O |
2.378(1) |
2.344(5) |
O |
575 |
|
Cs4Na2[Rh2(CO3)4Cl2]·8H2O |
2.380(2) |
2.601(3) |
Cl |
575 |
|
Na4[Rh2(SO4)4(H2O)2]·4H2O |
2.449(3) |
2.298(9) |
O |
580 |
|
trans-[Rh2(SO4)2(py)6]·6H2O |
2.604(2) |
2.25(1) |
N |
580,581 |
|
trans-[Rh2(SO4)2(py)6]·3H2O |
2.592(3) |
2.29(2) |
N |
580 |
|
Rh2(H2PO4)4(H2O)2 |
2.487(1) |
2.292(7) |
O |
584,585 |
|
trans-[Rh2(HPO4)2(py)6]·(pyH)2HPO4 |
2.598(2) |
2.226(7) |
N |
581,587 |
|
|
2.621(2) |
2.246(8) |
|
|
a Distances are given with up to 3 decimal digits.
Early reports of carbonato-bridged dirhodium compounds formulated as [C(NH2)3]2- Rh(CO3)2574 and [Rh2(CO3)4]4− 40,370noted that their properties resemble those of Rh2(O2CR)4L2 compounds but no specific structures were proposed. In particular, the [Rh2(CO3)4]4- species was prepared by two different methods: treatment of a Rh24+(aq) solution with an excess of CO32− ions followed by addition of alkali metal ions (Na+, K+, Cs+) to produce purple or dark-blue crystalline solids or, more directly by a ligand exchange procedure in which

528Multiple Bonds Between Metal Atoms Chapter 12
Rh2(O2CCH3)4 is heated in the presence of a concentrated aqueous solution of the alkali metal carbonate.40,41 The formulation of these compounds as salts of the [Rh2(CO3)4]4- ion was based on elemental analyses, infrared and electronic absorption spectra, magnetic susceptibility measurements and electrochemical data.40 The [Rh2(CO3)4]4- formulation was confirmed in 1980 when the X-ray crystal structure determinations of Cs4[Rh2(CO3)4(H2O)2]·6H2O and Cs4Na2- [Rh2(CO3)4Cl2]·8H2O were reported.575 The Rh–Rh bond lengths in these two compounds, 2.378(1) Å and 2.380(2) Å, respectively, are not significantly shorter than the corresponding distance in Rh2(O2CCH3)4(H2O)2 (2.386(1) Å),223 in contrast to the situation with dichromium compounds. The ax Cl− ligands in Cs4Na2[Rh2(CO3)4Cl2]·8H2O are associated with the Rh atoms at 2.60 Å, a distance that implies weak interactions; this is most likely due to the high negative charge of the [Rh2(CO3)4]4− unit.575 There is also evidence that, in aqueous solution, species such as [Rh2(HCO3)2]2+ may exist.40 Moreover, several bis-carbonate complexes that contain chelating 2,2'-bipyridine have been prepared from the formato complexes cis-Rh2(O2CH)2X2(bpy)2 X = Cl, Br, and characterized by infrared and electronic absorption spectroscopies.346
Prior to recognition of carbonate-bridged dirhodium compounds, there was evidence of sul- fate40,576-579 and acetate/sufate371 bridged species. Wilson and Taube reported that elution of the Rh24+(aq) ion from a cation exchange column with sulfate solutions allowed the isolation of a compound formulated as (NH4)4[Rh2(SO4)4].40 The structure of Na4[Rh2(SO4)4(H2O)2]·4H2O580 revealed great similarity to that of the tetrakis-carbonate complex, i.e., the two metal centers are bridged by four sulfate groups.580 Reaction of aqueous solutions of the sulfate compound with pyridine affords the crystalline hydrated adducts [Rh2(SO4)2(py)6]·nH2O, n = 3 or 6, with the bridging sulfate groups in a transoid arrangement,580,581 in contrast to the cisoid arrangement of the acetate groups in [Rh2(O2CCH3)2(py)6](CF3SO3)2.367,368 The relative ease of displacing the sulfate groups of [Rh2(SO4)4(H2O)2]4− is demonstrated by reaction of its Na+ salt with the nitriles RCN (R = CH3, C2H5 or C6H5) in the presence of CF3SO3H.582 In the case of CH3CN, this reaction affords the cation [Rh2(NCCH3)10]4+ (Section 12.4.2).
Treatment of Rh2(O2CCH3)4 with aqueous H3PO4 affords mixed acetate/phosphate species369 as well as Rh2(H2PO4)4(H2O)2,583 which exhibits the expected paddlewheel structure with ax water molecules.584,585 This compound gives acidic solutions when dissolved in H2O and reacts with pyridine to afford [Rh2(HPO4)2(py)6]·6H2O, (pyH)4[Rh2(HPO4)4(py)2] or [Rh2(HPO4)2(py)6]·(pyH)2HPO4, depending on the reaction conditions.581,584,586,587 The structure of [Rh2(HPO4)2(py)6]·(pyH)2HPO4581,587 reveals that the two HPO42− ligands are in a transoid arrangement similar to trans-[Rh2(SO4)2(py)6].580,581 Addition of a 40% HClO4 solution to [Rh2(HPO4)2(py)6]·(pyH)2HPO4 yields [Rh2(H2PO4)2(py)4(H2O)2](ClO4)2·2H2O.581 The electronic and CD spectra 588-590 of the tetraphosphate and tetrasulfate compounds have been reported.
12.4 Dirhodium Compounds with Unsupported Rh–Rh Bonds
12.4.1 The dirhodium(II) aquo ion
The dirhodium aquo species [Rh2(H2O)10]4+ was first generated591 according to the reaction:
2[Rh(H2O)5Cl]2+ + 2Cr2+(aq) Α [Rh2(H2O)10]4+ + 2CrCl2+(aq)
It is often written as [Rh2(H2O)10]4+ but the degree of hydration is speculative. The dimeric formulation of Rh24+(aq) is based on its cation exchange behavior, the similarity of its electronic absorption spectrum to that of Rh2(O2CCH3)4(H2O)2 and its diamagnetism, but attempts to

Rhodium Compounds 529
Chifotides and Dunbar
precipitate or crystallize it have failed to produce a solid form of [Rh2(H2O)10]4+.40,591,592 It has been shown, however, that a number of Rh(III) species can be employed as starting materials and that electron transfer reactions between Cr(II) and Rh(III) proceed through bridged transition states (inner-sphere mechanism).592
The reaction of Rh24+(aq) with O2 in a 2-3 M HClO4 solution is postulated to produce the purple paramagnetic superoxo complex [Rh2(O2−)(OH)2(H2O)n]3+.593,594 Treatment of [Rh2(H2O)10]4+ in an aqueous solution of HClO4 with NH4OH/NH3, py and/or en results in water exchange and formation of the corresponding [Rh2(H2O)10-m(base)n(OH)m](4-m)+ derivatives.595 Reaction of the latter with dioxygen affords superoxo and peroxo complexes, depending on the reaction conditions.595
An 17O NMR investigation of aqueous solutions of [Rh2(H2O)10]4+ revealed the presence of [Rh2(eq-H2O)8(ax-H2O)2]4+ and it was noted that the exchange of the two ax H2O ligands is at least a factor of 103 faster than the exchange of the eq-H2O groups at 298 K.596 The [Rh2(H2O)10]4+ ion decomposes within 1 h at 339 K forming metallic Rh and [Rh(H2O)6]3+.596
12.4.2 The [Rh2(NCR)10]4+ cations
In contrast to [Rh2(H2O)10]4+, the soluble organic analogs [Rh2(NCR)10]4+, R = CH3, CH3CH2, have been isolated by several independent routes and structurally characterized.582,597,598 One
method employs addition of excess Et3OBF4 to an acetonitrile solution of Rh2(O2CCH3)4(MeOH)2, which initially produces the purple complex cis-[Rh2(O2CCH3)2(NCCH3)6](BF4)2.361 Upon heating the reaction for several days, however, the purple colored solution turns orange and the dark orange-red homoleptic complex [Rh2(NCCH3)10](BF4)4 (Fig. 12.28) is isolated.597 Alternatively, Me3Si(CF3SO3) can be employed to remove the carboxylate ligands from the dinuclear unit as silylesters, or the acid HBF4·Et2O can be used to protonate them and liberate acetic acid, but the latter strategy leads to severe oiling problems which renders it less useful.598 The use of the more labile Rh2(O2CCF3)4 reduces considerably the reaction time. In addition, Rh2(O2CCH3)4, Na4[Rh2(CO3)4]·nH2O, or preferably Na4[Rh2(SO4)4(H2O)2]·4H2O can be heated in CH3CN/ CF3SO3H mixtures to afford [Rh2(NCCH3)8(H2O)2](PF6)4 which has been structurally characterized;599 when this compound is dissolved in CH3CN, it forms [Rh2(NCCH3)10](PF6)4,582 and many compounds of the type [Rh2(NCCH3)8L2](PF6)4 (L = DMF, DMSO, NH3, py, PPh3) depending on the identity of the solvent.582,600 Similar strategies have been employed to prepare [Rh2(NCEt)10](CF3SO3)4, [Rh2(NCC6H5)8L2](ClO4)4 (L = H2O or py),582 and [Rh2(NCEt)10](BF4)4, which has been the subject of single crystal X-ray studies.598 The structurally characterized cations [Rh2(NCR)10]4+ are listed in Table 12.7.
The Rh–Rh distances of the nitrile salts [Rh2(NCR)10]4+ are in the narrow range 2.604-2.625 Å, a fact which suggests that the Rh–Rh bond is not highly influenced by the ligand identity or the different counterions.598 Importantly, these nitrile Rh–Rh bonds are much shorter than in Rh2(dmg)4(PPh3)2 (2.936(2) Å)337,338 and the unsupported isocyanide compound [Rh2(CN-p-tol)8I2]2+ (2.785(2) Å),601 but longer than the average distance in tetracarboxylate systems (2.35-2.45 Å; Table 12.1). There is no significant repulsion between the essentially linear CH3CN molecules in [Rh2(NCCH3)10](BF4)4, contrary to Rh2(dmg)4(PPh3)2 (see Section 12.4.3), wherein the unfavorable interaction between the dmg groups induces considerable lengthening of the Rh–Rh bond.337,338 Therefore the cation [Rh2(NCCH3)10]4+ with a fully staggered (ρav = 44.8º) set of small eq ligands, which are neutral μ-donors, provides a reliable measure of the Rh–Rh bond distance in the absence of constraints or repulsions.
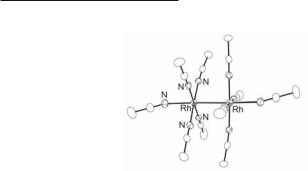
530Multiple Bonds Between Metal Atoms Chapter 12
Fig. 12.28. The cation in [Rh2(NCCH3)10](BF4)4. |
|
|
|
|
Table 12.7. Dirhodium compounds with unsupported Rh–Rh bonds |
|
|
||
Compound |
r (Rh–Rh)a (Å) |
r (Rh–Lax) (Å) |
Donor |
ref. |
|
|
|
atom(s) |
|
[Rh2(NCCH3)8(H2O)2](PF6)4·2H2O |
2.625(1) |
2.23(2) |
O |
599 |
[Rh2(NCCH3)10](BF4)4 |
2.624(1) |
2.191(5) |
N |
597,598 |
[Rh2(NCCH3)10](O3SCF3)4 |
2.616(2) |
2.15(1) |
N |
598 |
|
|
2.14(1) |
|
|
[Rh2(NCEt)10](BF4)4 |
2.604(1) |
2.180(6) |
N |
598 |
[Rh2(CN-p-tol)8I2](PF6)2 |
2.785(2) |
2.735(1) |
I |
601 |
Rh2(dmg)4(PPh3)2·H2O·C3H7OH |
2.936(2) |
2.430(5) |
P |
337,338 |
|
|
2.447(5) |
|
|
Rh2(dmg)4(py)2 |
2.726(1) |
2.219(8) |
N |
609 |
Rh2(hfacac)4(py)2 |
2.590(1) |
2.271(8) |
N |
582,610 |
|
|
2.245(8) |
|
|
Rh2(tmtaa)2 |
2.619(6) |
b |
b |
614 |
|
|
|||
Rh2(tmtaa)2·3C7H8 |
2.619(1) |
b |
b |
144 |
|
|
|||
[Rh2(pc)2(py)2]·2C6H6 |
2.741(2) |
2.309(8) |
N |
617 |
[Rh(CO)2Cl2]2[Rh2(quinCO)2(CO)2] |
2.671(1) |
2.522(2) |
Cl |
612 |
a Distances are given with up to 3 decimal digits. b No ax ligands.
A high-pressure 1H NMR study596 of CH3CN exchange kinetics for [Rh2(NCCH3)10]4+ supports the presence of [Rh2(eq-NCCH3)8(ax-NCCH3)2]2+ in solution. Consistently with the findings for [Rh2(H2O)10]4+ 596 and cis-[Rh2(O2CCH3)2(NCCH3)6]2+,364 the ax CH3CN ligands are more labile than those in eq positions. Lastly, the cation [Rh2(NCCH3)10]4+ has been reported to exhibit unusual spectroscopic properties, including reversible photochemical heterolytic cleavage of the Rh–Rh bond at η < 600 nm to yield the metastable photofragments [Rh(NCCH3)6]3+ and [Rh(NCCH3)4]+, which recombine to regenerate the original dimer in essentially quantitative yields.602,603 Exposure of [Rh2(NCCH3)10](BF4)4 to light after immobilization in ordered mesoporous silica promotes irreversible photodissociation to monomeric species.604
The mixed valence molecular wire [Rh(NCCH3)4(BF4)1.5] 598,605 is discussed with the rhodium blues in Section 12.5.2.
12.4.3 Complexes with chelating and macrocyclic nitrogen ligands
Compounds of this type that have been crystallographically determined are compiled in Table 12.7. The first example of a Rh24+ compound lacking bridging groups is Rh2(dmg)4(PPh3)2, which is prepared by reduction of Rh2(dmg)2Cl(PPh3) with an excess of NaBH4.606 Several

Rhodium Compounds 531
Chifotides and Dunbar
germane compounds with other ax donors (e.g., H2O, DMSO, py), prepared by a synthetic procedure starting from Rh2(O2CCH3)4, have been reported.607 The compound Rh2(dmg)4(PPh3)2 has a very long Rh–Rh distance of 2.936(2) Å.337,338 Despite the staggered conformation of the dmg moieties, they are prevented from bending back by the PPh3 ligands, thus repulsion between the dmg groups is primarily responsible for the long Rh–Rh distance.608 An appreciable shortening of the Rh–Rh bond by 0.21 Å takes place upon replacing the ax phosphine groups with pyridine to form Rh2(dmg)4(py)2.609 The estimated dissociation energy for homolytic cleavage of the Rh–Rh bond in Rh2(dmg)4(PPh3)2 is c. 20 kcal.mol−1.608
Heating aqueous solutions of Na4[Rh2(SO4)4(H2O)2]·4H2O with acetylacetone (acac) or hexafluoroacetylacetone (hfacac) to 80-90 °C under argon affords Rh2(acac)4 and Rh2(hfacac)4(H2O)2·2H2O with bidentate chelating diketonate groups.582 In the pyridine adduct Rh2(hfacac)4(py)2, the Rh–Rh bond of 2.590(1) Å582,610 is shorter by c. 0.10 Å than that in Rh2(dmg)4(py)2, a fact which clearly shows the effect of the bulky dmg groups on the Rh–Rh bond length. The chelate rings are not eclipsed, instead they are twisted by c. 42° with respect to each other. Reaction of Rh2(hfacac)4L2, L = H2O, py with PPh3 in toluene, in the absence of O2, is believed to form the paramagnetic species Rh(hfacac)2(PPh3),600 whereas in the presence of O2, it produces the peroxo complex (Ph3P)(hfacac)2Rh(µ-O2)Rh(hfacac)2(PPh3).611
A related compound is the tetranuclear acylrhodium complex [Rh(CO)2Cl2]2[Rh2- (quinCO)2(CO)2] (quinCO: 8-quinoline acyl), which is formed by reacting [(CO)2RhCl]2 with 8-quinoline carboxaldehyde. The rather short unsupported Rh–Rh bond of 2.671(1) Å is mostly attributed to stacking interactions between the acylquinoline ligands.612
The macrocycle tetraaza[14]annulene, abbreviated H2tmtaa (12.39), has a saddle-shaped conformation resulting from internal steric constraints which cause displacement of the coordinated metal from the N4 plane.613,614 This makes it possible for this 14-membered ring, which is an anti-aromatic system (4n), to coordinate in a tetradentate fashion to metal atoms that participate in metal-metal bonds. The reaction of H2tmtaa with Rh2(O2CCH3)4 in EtOH615 affords Rh2(tmtaa)2 with a Rh–Rh distance of 2.619(1) Å144,614 (there is an earlier report quoting a similar Rh–Rh distance of 2.625 Å,616 but full crystallographic details were not given). The shorter Rh–Rh bond distance compared to Rh2(dmg)4(py)2 (2.726(1) Å)609 and [Rh2(CN-p-tol)8I2](PF6)2 (2.785(2) Å)601 is attributed to the absence of ax ligands, whereas the longer Rh–Rh distance compared to Rh2(hfacac)4(py)2 (2.590(1) Å), is likely a result of the 0.219 Å displacement from the N4 plane.144 The red diamagnetic di(pyridine)phthalocyaninatorhodium(II) complex, [Rh(py)(pc)]2 (Fig. 12.29a) is another example of a dirhodium unit with a macrocycle, namely phthalocyanine (H2pc; 12.40), which has been structurally characterized.617 The cofacial rhodium phthalocyaninate units have a Rh–Rh distance of 2.741(2) Å, which is comparable to that in Rh2(dmg)4(py)2 (2.726(1) Å)609 and suggests a strong unsupported Rh–Rh single bond. The pc-pc repulsions are minimized by their staggered conformation with a twist angle of 42° (Fig. 12.29b).617
Porphyrins (12.41) are 16-membered aromatic rings (4n + 2) that coordinate to metals in a tetradentate fashion. Dirhodium compounds with the tetraphenylporphyrin (TPP) and octaethylporphyrin (OEP) dianions have been extensively studied due to their rich radical chemistry and potential importance in biologically relevant processes. In the case of OEP, the Rh(III) hydrido complex Rh(OEP)H is converted, upon heating, to the dimeric complex [Rh(OEP)]2, which has a Rh–Rh bond.618,619 The reaction of [Rh(OEP)]2 with O2 at -80 °C affords the paramagnetic Rh(OEP)(O2) which, upon warming to room temperature, produces a peroxo species formulated as [Rh(OEP)]2(O2).620,621 A paramagnetic mononuclear compound formulated as [Rh(TPP)] has been prepared by reaction of [Rh(CO)2Cl]2 with a solution of H2TPP in refluxing acetic acid.622 The similar behavior of ‘Rh(TPP)’ to Rh(OEP)(O2) suggested that it may actually

532Multiple Bonds Between Metal Atoms Chapter 12
be Rh(TPP)(O2); this is further supported by the fact that its sublimation in vacuum affords a diamagnetic compound consistent with the formula [Rh(TPP)]2, which reacts with O2 and NO similarly to [Rh(OEP)]2.620 The Rh–Rh bond dissociation energy of [Rh(OEP)]2 has been estimated c. 16.5 kcal mol−1 by 1H NMR line broadening measurements, but [Rh(OEP)]2 and [Rh(TPP)]2 have not been crystallographically characterized.623,624 The radical-like reactivity of [Rh(OEP)]2, initiated by dissociation of [Rh(OEP)]2 to [Rh(OEP)], has been extensively studied.625-633 The reactions of the dirhodium diporphyrin complexes Rh2(DPB)634 (DPB: diporphyrinatobiphenylene) and [Rh(OMP)]2635 (OMP: 2,3,7,8,12,13,17,18-octamethoxyporphyrin dianion), which have a Rh–Rh single bond, with various molecules have been studied.
12.39 |
12.40 |
12.41 |
Fig. 12.29. The structure of the phthalocyanine (H2pc) dirhodium complex [Rh(pc)(py)]2; (a) side view, (b) view along the Rh–Rh axis.
For tetramesitylporphyrin (H2TMP), the mononuclear radical [Rh(TMP)]• is the stable form since ligand steric requirements preclude Rh–Rh bonding.636,637 The focal point of subsequent studies has been the design of a series of tethered diporphyrin ligands, by linking two sterically demanding TMP derivatives with a series of diether spacers, in order to improve the kinetics and retain the selectivity of the stable bimetalloradical •Rh(porphyrin)-X-(porphyrin)Rh• (X = spacer) reactions with substrates such as H2 and CH4.638,639
Although the Schiff base H2salen (12.42) and the sulfur-based ligand ‘But-H2S4’ (12.43; ‘But- H2S4’: 1,2-bis(2-mercapto-3,5-di-But-phenylthio)ethane) are not macrocycles, they are mentioned here for practical purposes. Compounds formulated as Rh2(salen)2(py)2,640 [Rh(salen)]2 and a few related derivatives with other Schiff bases have been reported but not structurally

Rhodium Compounds 533
Chifotides and Dunbar
characterized.641 The chelating ligand ‘But-H2S4’ (12.43) is coordinated in a tetradentate fashion in Rh2(But-S4)2;642 each thiolate group of the ligand bridges two rhodium centers.
12.42 |
12.43 |
12.5 Other Dirhodium Compounds
12.5.1 Complexes with isocyanide ligands
Although Rh(I) isocyanide complexes were known, it was not until much later that Rh24+ isocyanide species were obtained by reaction of [Rh(CNR)]+ and [Rh(CNR)4X2]+ (X = halide) in solvents of high dielectric constants, or by reaction of I2 with [Rh(NCR)4]+ in a 1:2 mole ratio.643,644 A species with a Rh–Rh bond, ax Rh-X bonds and no bridging groups is favored for the products [Rh2(CNR)8X2]2+ and confirmed by the crystal structure determination of [Rh2(CN-p-tol)8I2](PF6)2.601 A class of germane complexes with di-isocyanide ligands are formulated as [Rh2(LL)4X2]2+, LL: 1,3-di-isocyanopropane (abbreviated bridge, 12.44),645-648 2,5- di-isocyano-2,5-dimethylhexane (abbreviated TMB, 12.45),648,649 and 1,8-di-isocyanomenthane (abbreviated dimen, 12.46).650 Salts of the [Rh2(LL)4X2]2+ cations (X = Cl, Br or I) are prepared by oxidation of [Rh2(LL)4]2+ with molecular halogens.648,650
12.44 |
12.45 |
12.46 |
The structures of the isocyanide complexes [Rh2(bridge)4Cl2]Cl2647 and [Rh2(TMB)4Cl2]- (PF6)2649 have been crystallographically determined (Table 12.8). Although the Rh–Rh bond lengths in these compounds (2.837(1) and 2.770(3) Å, respectively) are similar to that in [Rh2(CN-p-tol)8I2](PF6)2 (2.785(2) Å),601 the C–Rh–Rh–C torsion angles vary considerably. The isonitrile groups in [Rh2(bridge)4Cl2]Cl2 are eclipsed, whereas in [Rh2(TMB)4Cl2](PF6)2 and [Rh2(CN-p-tol)8I2](PF6)2, they are twisted by 33° and 28-35°, respectively, from the fully eclipsed geometry.649
Table 12.8. Other dirhodium compounds
Compound |
|
r (Rh–Rh)a |
r (Rh–Lax) |
Donor |
ref. |
|
(Å) |
(Å) |
atom(s) |
|
|
|
Rhodium blues |
|
|
|
|
H3[Rh4{CN(CH2)3NC}8Cl][CoCl4]4·6H2Ob |
|
2.932(4)c |
2.613(8) |
Cl |
653 |
|
|
2.923(3)c |
2.643(9) |
|
|
|
|
2.775(4)d |
|
|
|

534Multiple Bonds Between Metal Atoms Chapter 12
Compound |
r (Rh–Rh)a |
r (Rh–Lax) |
Donor |
ref. |
|
(Å) |
(Å) |
atom(s) |
|
{Rh(NCCH3)4](BF4)1.5} e |
2.928(1) |
|
|
598,605 |
|
2.844(1) |
|
|
|
{[Rh(µ-pz)(CNBut)2]4}(PF6)2 |
2.721(4)f |
|
|
673 |
|
2.723(4)f |
|
|
|
|
2.713(4)g |
|
|
|
[Rh4(µ-O2CH)4(bpy)4](PF6)2 |
2.668(1)h |
|
|
235 |
|
2.780(1)i |
|
|
|
{[Rh4(O2CH)4(bpy)4]BF4·0.5C4H8O2} j |
2.678(3)h |
|
|
358 |
|
2.733(4)i |
|
|
|
|
2.921(3)k |
|
|
|
{[Rh2(µ-O2CCH3)2(bpy)2]BF4·H2O} e |
2.666(2)l |
|
|
678 |
|
2.833(2)m |
|
|
|
{[Rh2(µ-O2CCH3)2(phen)2]PF6·0.5Me2CHOH} e |
2.652(1)n |
|
|
677 |
|
2.739(1)o |
|
|
|
|
2.832(1)o |
|
|
|
[Rh3(CNCH2Ph)12I2]Br3p |
2.785(2) |
2.735(1) |
I |
675 |
[Rh3(s-pqdi)4(pqdi)2]Cl·3DMF |
2.754(2)q |
|
|
676 |
Isocyanide compounds |
|
|
|
|
[Rh2{CN(CH2)3NC}4Cl2]Cl2·8H2O |
2.837(1) |
2.447(1) |
Cl |
647 |
[Rh2(TMB)4Cl2](PF6)2 |
2.770(3) |
2.425r |
Cl |
649 |
[Rh2(CN-p-tol)8I2](PF6)2 |
2.785(2) |
2.735(1) |
I |
601 |
trans-[Rh2(µ-pz)(CNBut)2(dppm)2Cl2]PF6 |
2.768(1) |
2.489(1) |
Cl |
662 |
|
|
2.471(1) |
|
|
trans-[Rh2(µ-pz)(CNBut)2(dppm)2I2]BF4 |
2.829(3) |
2.738(3) |
I |
663 |
|
|
2.766(6) |
|
|
{[Rh2(µ-pz)2(I)(CNBut)4]2(µ-I)}CF3SO3 |
2.632(1) |
2.728(1) |
I |
485 |
|
|
2.754(1) |
|
|
|
|
2.790(1) |
|
|
|
|
2.812(1) |
|
|
a Distances are given with up to 3 decimal digits. |
|
|
|
|
b |
Rh(I)Rh(II)Rh(II)Rh(I) chain with an average oxidation state of +1.5 for each of the four Rh atoms in the |
|
tetranuclear unit. |
c |
Rh(I)···Rh(II) distance within the binuclear unit. |
d |
Rh(II)···Rh(II) distance between the two binuclear units Rh23+. |
e |
Mixed valence Rh(I)–Rh(II) infinite wire. |
f |
Rh···Rh distance within [Rh2(pz)2(CNBut)4]+ unit. |
g |
Rh···Rh separation between [Rh2(pz)2(CNBut)4]1+ units. |
hRh–Rh distance for entity supported by the formato group within the tetranuclear linear [Rh4(µ-O2CH)4(bpy)4]+ unit.
i |
Rh···Rh interaction within the tetranuclear linear [Rh4(µ-O2CH)4(bpy)4]+ unit. |
j |
Infinite rhodium wire with each Rh atom in the +1.25 oxidation state. |
k |
Rh···Rh interaction between the tetranuclear linear [Rh4(µ-O2CH)4(bpy)4]+ units. |
l |
Rh–Rh distance within [Rh2(µ-O2CCH3)2(bpy)2]+ units. |
m Rh···Rh distance between [Rh2(µ-O2CCH3)2(bpy)2]+ units. |
|
n |
Rh–Rh distance within [Rh2(µ-O2CCH3)2(phen)2]+ units. |
o |
Rh···Rh separations between [Rh2(µ-O2CCH3)2(phen)2]+ units. |
p |
The molecule contains nearly linear I–Rh–Rh–Rh–I units. |
q |
Average distance. |
r |
Esd not reported. |