

Запитання для самоконтролю
1. Зобразіть графічно характер залежності поверхневого натягу від концентрації розчину поверхнево-активної речовини.
2. Дайте графічне зображення ізотерми адсорбції і поясніть явища, що відбуваються на поверхні адсорбенту, коли адсорбція максимальна.
3. Дайте аналіз адсорбційного рівняння ізотерми Ленгмюра і поясніть фізичний зміст констант цього рівняння.
4. Які види адсорбції можуть бути на поверхні твердого тіла, що занурене в розчин?
5. Наведіть приклади адсорбції газів. Опишіть і намалюйте розміщення поверхнево-активних молекул на поверхні рідин.
6. Наведіть приклади йонообмінної адсорбції в природі і в техніці.
7. Як можна пояснити легке вимивання розчинних речовин піщаних грунтів і яке це має значення для родючості грунтів?
8. Поясніть суть хроматографічного методу аналізу Цвета і наведіть приклади застосування цього методу.
9. У чому полягає правило вирівнювання полярностей Ребіндера?
10. Для яких видів фізичної адсорбції цей процес може бути необоротним?
11. В чому полягає полімолекулярна адсорбційна теорія БЕТ? Приведіть основні типи ізотерм адсорбції.
Тема 6. Фізико-хімія дисперсних систем та розчинів високомолекулярних сполук
Основи термодинаміки дисперсних систем. Дисперсними називають системи, які складаються з великої кількості диспергованих частинок – дисперсної фази, розподілених у рідкої, твердої або газовій фазах – дисперсійного середовища.
Дисперсні системи можуть бути одержані або диспергуванням твердих речовин, або утворенням твердої фази із розчинів (конденсаційно кристалізаційні і коагуляційні структури).
Розміри і форма часточок визначально впливають на властивості дисперсних систем. У більшості випадків утворення дисперсних систем потребує витрати роботи: або у вигляді механічної, або за рахунок процесів, що відбуваються в самій системі (в тому числі хімічних). Дисперсні системи, що виникли таким чином, є термодинамічно нерівноважними. Для свого скільки-небудь тривалого існування вони потребують спеціальної стабілізації. Такі термодинамічно нерівноважні дисперсні системи називають ліофобними.
На відміну від ліофобних існує клас дисперсних систем, які виникають внаслідок самовільного диспергування макроскопічної фази, тобто без витрати механічної роботи, що підводиться зовні. Такі системи називають ліофільними. Вони є термодинамічно рівноважними і не потребують будь-якої додаткової стабілізації.
Виникнення дисперсної системи внаслідок утворення частинок, що зародилися у новій стабільній фазі, можливе в любій метастабільній системі. Метастабільність, яка пов’язана з віддаленням від меж рівноважних умов існування даної системи, може бути викликана як відхиленням в хімічному складі фаз (пересичення), так і внаслідок фізико-хімічного впливу на систему (зміна тиску або температури).
|
|
|
Рис.6.1. Схема самовільного утворення ліофільної дисперсної системи |

Розглянемо процес утворення дисперсної системи шляхом диспергування макрофаги на прикладі відщеплення одної сферичної частинки радіусом r від макрофаги І, яка знаходиться в області стабільності цієї фази (рис.6.1). На рисунку цьому процесу відповідає перехід від стану І до стану ІІІ. Роботу утворення (диспергування) частинки (Wd) можна виразити як:
![]() (6.1)
(6.1)
де SK – поверхня критичного зародку.
Різниця
між хімічними потенціалами речовини
частинки
![]() і вихідної стабільної фази
і вихідної стабільної фази
![]() в цьому випадку складає:
в цьому випадку складає:
![]() ,
(6.2)
,
(6.2)
де N – кількість речовини (в молях), що утворює зародок; V – молярний об’єм речовини в зародку.
Утворення сферичної частинки (ІІІ) можливе також із деякої макроскопічної фази ІІ, стан якої відрізняється від стану макрофаги.
Їх хімічні потенціали різні, тому
![]() .
.
Робота
утворення цієї частинки, яка пов’язана
зі зміною фазового стану
![]() ,
дорівнює:
,
дорівнює:
![]() .
.
В той же час
![]() ,
,
де
![]() - “хімічна” робота
- “хімічна” робота
![]() переносу N речовини із стану ІІ в стабільну
фазу І:
переносу N речовини із стану ІІ в стабільну
фазу І:
![]() (6.3)
(6.3)
Таким чином, робота утворення сферичної частинки при фазовому перетворенні має вираз:
![]() (6.4)
(6.4)
З цього
виразу випливає, що величина роботи
![]() утворення частинки дисперсної фази з
макрофаги іншого агрегатного стану або
хімічного складу відрізняється від
величини роботи утворення частинок із
того ж агрегатного стану і складу
утворення частинки дисперсної фази з
макрофаги іншого агрегатного стану або
хімічного складу відрізняється від
величини роботи утворення частинок із
того ж агрегатного стану і складу
![]() на величину “хімічної” роботи
на величину “хімічної” роботи
![]() ,
яка залежить від різниці хімічних
потенціалів речовини в макроскопічних
фазах І і ІІ і пов’язаної з енергією
фазового переходу.
,
яка залежить від різниці хімічних
потенціалів речовини в макроскопічних
фазах І і ІІ і пов’язаної з енергією
фазового переходу.
У випадку рівності хімічних потенціалів:
![]() (6.5)
(6.5)
вираз (6.4) приймає вигляд:
 (6.6)
(6.6)
Цей вираз вперше отримав Гіббс. Як бачимо з виразу:
![]()
тоді
як
![]() компенсуються “хімічною” роботою
компенсуються “хімічною” роботою
![]() ,
а рівність (6.5) є нестійкою рівновагою
в умовах якої відбувається виникнення
критичних зародків нової фази.
,
а рівність (6.5) є нестійкою рівновагою
в умовах якої відбувається виникнення
критичних зародків нової фази.
Колоїдно-дисперсні системи. Мікрогетерогенні (колоїдні) системи займають проміжковий стан між макроскопічними гетерогенними системами і молекулярними розчинами – гомогенними системами.
Колоїдно-дисперсні системи характеризуються
розміром частинок дисперсної фази. Чим
дрібніші часточки і чим більше вони
витягнуті, тим сприятливіші умови для
виникнення міжчасткових контактів і
колоїдного структуроутворення. Проте
диспергування зумовлює надзвичайно
велику вільну поверхневу енергію
часточок. Залежність сумарної поверхневої
енергії (![]() )
від ступеня дисперсності (
)
від ступеня дисперсності (![]() )
часточок має екстремальний характер
(рис.6.2). Ступінь дисперсності, що
відповідає інтервалу
)
часточок має екстремальний характер
(рис.6.2). Ступінь дисперсності, що
відповідає інтервалу
![]() від 7,0 до 9,2, називається колоїдним
ступенем дисперсності. Слід відмітити,
що часточки з дуже великим ступенем
дисперсності при подальшому зменшенні
їх розміру, зменшують сумарну поверхневу
енергію (права частина залежності
від 7,0 до 9,2, називається колоїдним
ступенем дисперсності. Слід відмітити,
що часточки з дуже великим ступенем
дисперсності при подальшому зменшенні
їх розміру, зменшують сумарну поверхневу
енергію (права частина залежності
![]() від
від
![]() ,
рис.6.2).
,
рис.6.2).
|
|
|
Рис. 6.2. Залежність сумарної поверхневої енергії 1 м3 речовини від ступеня дисперсності (заштрихована частина – колоїдна ступінь) |

Класифікація дисперсних систем. В основі класифікації дисперсних систем лежать три загальні ознаки: дисперсність, гетерогенність та питома поверхня.
За ступенем дисперсності дисперсні системи поділяють на три типи: грубодисперсні (макрогетерогенні і мікрогетерогенні), колоїдно-високодисперсні і молекулярно-дисперсні (табл.6.1). Цей поділ зумовлений розмірами частинок дисперсної фази.
Класифікація дисперсних систем за агрегатним станом фаз (дисперсної фази і дисперсійного середовища) охоплює більшу частину різноманіття колоїдів і дає змогу виділити 9 (або строго кажучи, 8) типів дисперсних систем (табл.6.2). В неї агрегатні стани фаз позначають літерами: Т – тверда; Р – рідка; Г – газоподібна, а дисперсні системи – як дроби, де чисельник – дисперсна фаза, знаменник – дисперсійне середовище. У випадку, коли агрегатні стани фаз однакові до літер дробу добавляють індекси 1 – агрегатний стан дисперсної фази, а 2 – дисперсійного середовища (Т1/T2, P1/P2).
З наведеної класифікації (табл.6.2) випливає, що дисперсні системи, в яких дисперсійне середовище газоподібне або рідке, за кінетичними властивостями частинок дисперсійної фази можна віднести до вільно дисперсних систем (золі), тобто у яких дисперсна фаза рухома. До них відносяться аерозолі, емульсії, ліозолі. Дисперсні системи з твердим дисперсійним середовищем, у якому частинки дисперсної фази можуть вільно пересуватися, а також висококонцентровані суспензії (бетонне тісто, глиняні пасти) і піни відносяться до зв’язанодисперсних систем (драглі). Зв’язанодисперсні системи містять у своїй структурі мікропори (2 - 200 нм) і макропори (порожнечі, канали) (> 200 нм), тобто являють собою високопористі тіла.
Таблиця 6.1
Класифікація дисперсних систем залежно від
розмірів частинок дисперсної фази
|
Тип системи |
Дисперсність |
Розмір частинок, м |
Властивості та приклади |
|
Грубодисперсні: макрогетерогенні
мікрогетерогенні |
102 - 104
104 – 107 |
10-2– 10-4
10-4 – 10-7 |
Частинки, видимі неозброєним оком. Курява, хмари, кіптява, туман, дим. Частинки, видимі у звичайний мікроскоп. Емульсії, піна, грунт |
|
Колоїдно-високодисперсні (ультрамікро-гетерогенні) |
107 – 109 |
10-7 – 10-9 |
Частинки, видимі тільки в ультра- і електронний мікроскоп, опалесцен-ціюють. Золі, сік рослин, молоко, плазма |
|
Молекулярно-(йонно)-диспесрні |
Менша за 109 |
Менший за 10-9 |
Молекули та йони, невидимі в ультра-мікро-скопі, прохо-дять ультра-фільтри. Істин-ні розчини (цукру, спирту, солей, кислот, основ, тощо) |
Таблиця 6.2
Класифікація дисперсних систем за агрегатним станом фаз
|
Умовне позна-чення системи |
Диспер-сійне середо-вище |
Дисперсна фаза |
Назва системи та приклади |
|
T1/T2 |
Тверде тіло |
Тверде тіло |
Мінерали, сплави, тверді золі, солідозолі, емалі |
|
P/T |
-“- |
Рідина |
Вологий грунт, тканини клітин живих організмів, рослин, віск, бурштин, перли |
|
Г/T |
-“- |
Газ |
Пінобетон, пінопласт, адсорбенти, цеоліти, фомалюм, туф |
|
T/P |
Рідина |
Тверде тіло |
Суспензії, пасти, ліозолі (золі) |
|
P1/P2 |
-“- |
Рідина |
Емульсії (молоко, латекси, сира нафта) |
|
Г/P |
-“- |
Газ |
Піни (кипляча вода) |
|
T/Г |
Газ |
Тверде тіло |
Аерозолі (дим, пил, порошки, кіптява, перісті хмари) |
|
Р/Г |
-“- |
Рідина |
Аерозолі, туман, хмари (кучові) |
|
- |
-“- |
Газ |
Мікронеоднорідні гомогенні системи, що обумовлені флуктуацією густини (атмосфера) |
Колоїдно-дисперсні системи з рідким дисперсійним середовищем (ліозолі) називають колоїдними розчинами або просто золями. Якщо дисперсійним середовищем є вода такі золі називаються гідрозолями, а у випадку органічних рідин – органозолями, тобто якщо спирт – алкозолем, якщо етер – етерозолем і т.п.
Необхідною умовою утворення дисперсних систем типу Р1/Р2, які називаються емульсіями, є їх утворення із взаємно нерозчинних або малорозчинних рідин.
Дисперсні системи типу Г1/Г2 відсутні. Проте навіть в цьому випадку слід приймати до уваги специфічну мікронеоднорідність суміші газів, що обумовлена флуктуацією густини. Саме наявністю флуктуації густини і розсіюванням на неї світла пояснюється голубий колір неба: якби атмосфера була зовсім однорідною („оптично порожньою”), то небо було б чорним.
В дисперсних системах частинки однієї дисперсної фази можуть мати міцний зв’язок з дисперсійним середовищем, а другої – слабкий. За цією ознакою колоїдно-дисперсні системи поділяються на два класи: ліофільні, колоїдні частинки які значно взаємодіють з дисперсійним середовищем, та ліофобні, колоїдні частинки які взаємодіють мало або практично зовсім не взаємодіють. Ліофільні колоїдно-дисперсні системи, дисперсійним середовищем яких є вода, називають гідрофільними, а ліофобні – гідрофобними. Типовими прикладами гідрофільних колоїдних систем є суспензії глини (золі гідроксидів металів), а гідрофобних – колоїдні розчини золота (Ауруму), сульфідів металів.
Ліофільні системи утворюються шляхом довільного диспергування, отже, вони є термодинамічно стійкими.
Ліофобні системи утворюються з перенасичених систем або шляхом диспергування великих частинок, внаслідок чого вони термодинамічно нестійкі. Тому гідрофільні золі добувають у значно більших концентраціях ніж гідрофобні; вони стійкіші до дії електролітів. Осади, які утворюють гідрофільні золі, дуже пухкі, об’ємні, що обумовлюються наявністю між частинками прошарків дисперсійного середовища внаслідок слабкої взаємодії між частинками.
За топографічною ознакою частинок дисперсної фази дисперсні системи поділяються на корпускулярні, ламінарні (плівкові), фібрілярні (волокнисті).
Наведена сукупність класифікацій надає можливість більш повніше характеризувати любу дисперсну систему, що розглядається, чого неможливо досягти при використанні любої окремої класифікації.
Високомолекулярні сполуки та їх властивості. Високомолекулярні сполуки (ВМС), або полімери, містять у складі своїх молекул десятки і сотні тисяч (мільйонів) атомів. Побудовані вони з елементарних ланок, які повторюються, внаслідок взаємодії і з’єднання між собою однакових або різних молекул-мономерів. ВМС являють собою групу органічних речовин з специфічними властивостями, які притаманні тільки їм. Так, ВМС здатні до утворення термодинамічно рівноважних молекулярних розчинів з особливими термодинамічними властивостями, що обумовлені гнучкістю ланцюгів макромолекул, які володіють великою кількістю конформацій.
В розчинах, в залежності від характеру взаємодії макромолекул, ВМС можуть існувати або у вигляді гнучких ланцюгів (статистичних клубків), або як щільні глобули згорнутих ланцюгів, або у вигляді асоціатів одної молекули з іншою. Встановлено, що розчини високополімерів гомогенні, тобто мають властивості справжніх розчинів низькомолекулярних сполук. Проте за величиною молекул розчини полімерів наближаються до колоїдних. Тому розчини ВМС мають деякі ознаки колоїдних розчинів.
Синтетичні і природні ВМС. ВМС утворюються полімеризацією або поліконденсацією мономерів.
Полімеризація відбувається з мономерами, які містять атоми з ненасиченими кратними зв’язками (СН2 = СН2, СН2 = С – СН = СН2).
│
СН3
При цьому подвійний зв’язок між атомами кожної з молекул мономеру розривається і за рахунок таких зв'язків відбувається сполучення величезної кількості (n) молекул мономеру у великі молекули полімеру:
n .
СН2 = С – СН = СН2
![]() ( - СН2 - С = СН – СН2 - )n.
( - СН2 - С = СН – СН2 - )n.
│ │
СН3 СН3
(ізопрен) (поліізопрен (каучук))
Поліконденсація супроводжується виділенням низькомолекулярних речовин:

(терефталева кислота) (етиленгліколь)
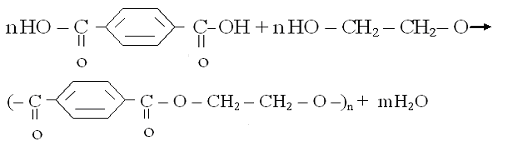
(поліетилентерефталат)
Можливо також утворення нових високомолекулярних сполук з мономерів, внаслідок їх хімічного перетворення:

Можливі оцінки розчинності полімерів, взаємодії полімеру з розчинником і визначення межи розчинності до сих пір ще достатньо не вивчені. Встановлено, що хімічна і структурна спорідненість між полімером і розчинником полегшує розчинність. Високомолекулярні сполуки можуть утворювати і колоїдні розчини (дисперсії). В таблиці 6.3 приведені деякі представники полімерів і речовини, що їх розчинюють.
В залежності від природи полімеру і розчинника і термодинамічних умов розчини полімерів в низькомолекулярних рідинах розглядають або як колоїдні системи, або як істинні розчини.
ВМС, що утворені з двох-трьох і більше ланок різного складу, поєднують у собі властивості вихідних мономерів. Їх називають сополімерами. Прикладами сополімерів є сополімери стиролу з малеїновою кислотою
|
( ─ СН2 – С = СН – СН2 ─) n ,
С6Н5 |
сополімери стиролу з бутадієном і акрилонітрилом
|
( ─ СН2 – СН – СН2 ─ СН = СН – СН2 – СН2 – СН ─) n ,
С6Н5 CN |
Таблиця 6.3
Високомолекулярні сполуки і речовини, що їх розчинюють
|
Полімер |
Формула елементарного ланцюга полімеру |
Розчинники |
|
|
Поліакриламід |
- СН2 – СН - │ О = С – NH2 |
Вода |
|
|
Полідиметил-силоксан |
- O – Si(CH3)2 - |
Вода |
|
|
Поліізобутилен |
- CH2C(CH3)2 - |
Бензен |
|
|
Поліетиленоксид |
- CH2CH2O - |
Хлороформ |
|
|
Полівінілацетат |
- CH2CH(OCOCH3) - |
Ацетон, діоксан, бензен |
|
|
Полівінілхлорид |
- CH2CHCl - |
Циклогексанон, хлорбензен |
|
|
Полістирол |
- CH2CH(C6H5) - |
Бензин, етилацетат, метилетилкетон |
|
|
Поліметилмета-крилат |
- CH2C(CH3)(COOCH3) - |
Ацетон, бензен, хлороформ |
|
|
Триацетат целюлози |
- (C6H7O2)(OOCCH3)3 - |
Оцтова кислота |
|
|
Карбаміднофор-мальдегідна смола |
- N(CONH2)CH2N(CONH2)CH2 - |
Етанол, вода |
|
|
|
|||
сополімери аценафтилену з вінілкарбазолом, стиролом, метакрилатом, вінілацетатом, ізобутиленом та ін. Їх застосовують в якості термостійких йоннообмінних смол, плівок, волокон, штучного грунту тощо.
За допомогою ультразвукових коливань, електромагнітних вібраторів та іншими способами, ряд механічно подрібнених полімерів утворюють з ланцюгів своїх молекул радикали. Взаємодія між такими радикалами надає можливість одержання нових високополімерів.
Високомолекулярні сполуки можуть мати як органічну, так і неорганічну природу.
Для утворення ланцюгів полімерних сполук можуть служити не тільки Карбон або Силіцій, але і Алюміній, Бор, Магній, Фосфор, Тітан та ін. Наприклад, це поліфосфорні кислоти:

фосфоноксилоксани:


поліфосфіноборани:
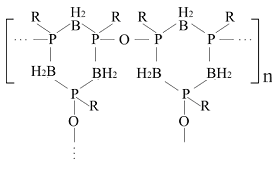
гідраргіліт:
 .
.
Значну кількість природних високомолекулярних сполук біологічного походження (вуглеводи, білки) також відносять до полімерів (біополімерів), наприклад целюлозу, крохмаль, полісахариди:

|
каучук |
|

Клітини усіх тканин живих організмів і рослин та міжклітинна речовина також є біополімерами.
Якщо полімери в основному або бічних ланцюгах містять йоногенні групи, їх називають поліелектролітами. До них відносяться желатина, казеїн, альбумін і альгінати. З синтетичних поліелектролітів до них відносяться поліакрилова кислота, поліакриламід і інші полімери, які містять кислотні
(─CОО-, HOSO3-, ОРО3Н2-) або основні (NH3+, NHC(NH2)2+, N(CH3)3+) групи.
До полікислот відносяться нуклеїнові кислоти, більшість муколісахаридів, гіалуронова, альгінова і карагінова кислоти, гуміарабік.
Найпростіша синтетична поліакрилова полікислота у водному розчині слабко дисоціює:
![]()
Типовим представником поліоснов є полівініламін, йоннізація якого в кислому середовищі відбувається із захопленням протону:

Поліоснови, як і полікислоти сильніше йонізовані у сольовій формі:

Сполуки в яких в одному ланцюгу сполучаються кислотні і основні групи називаються поліамфолітами, наприклад:

До поліамфолітів відносяться усі білки. Поліамфоліти грають велику роль в природі: в склад біологічних мембран входять ліпіди, які містять амінні і кислотні залишки, що визначають їх амфотерні властивості.
Основні особливості поліелектролітів зводяться до наступного. У воді їх макромолекули обмінюють протони і інші йонногенні групи на йони оточуючого середовища і таким чином набувають заряд. Так карбоксили полікислот в кислотній області рН залишаються практично нейоннізованими, а в нейтральній або лужній області з’являються заряджені групи – СОО-. Поліоснови, навпаки, залишаються нейонізованими в лужному середовищі і набувають позитивний заряд (NH3+) в кислому. Залежність заряду поліелектроліту від рН середовища представлена на рисунку 6.3.
|
а) |
б) |
|
Рис.6.3. Залежність заряду поліелектроліту від рН розчину: а) 1 – поліелектроліт, що містить тільки катіоногенні групи; 2 – поліелектроліт, що містить аніоногенні функціональні групи; б) – амфотерний поліелектроліт |
|


У водних розчинах поліамфоліти дуже легко гідролізуються за рівнянням:

Основні
групи – NH3ОН
в молекулі
![]() дисоціюють слабкіше ніж кислотні –
СООН,
тому молекула білка має негативний
заряд:
дисоціюють слабкіше ніж кислотні –
СООН,
тому молекула білка має негативний
заряд:
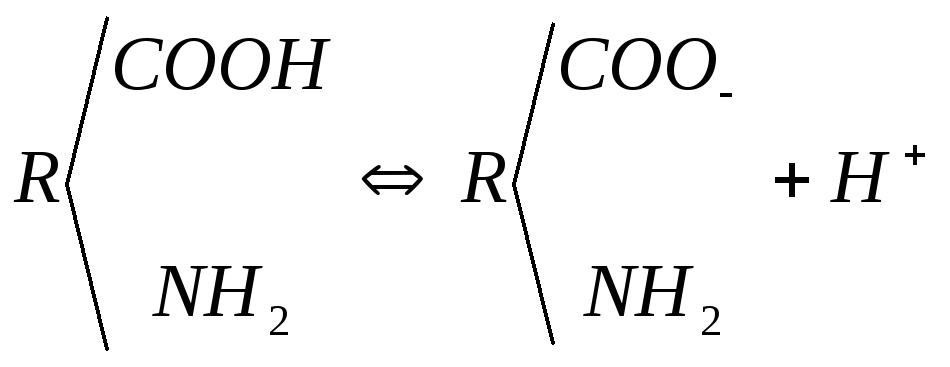
В кислому середовищі білкові молекули заряджаються позитивно:

Таким чином, знак зарядів амфотерних молекул білка залежить від реакції розчину (рис.6.3, б). Як видно з рисунка, точка перетину кривої з пунктирною лінією, відповідає нейтральному стану макромолекули в розчині. Такий ізоелектричний стан (рІ) у різних білків настає при різних концентраціях йонів Гідрогену розчину (табл.6.4).
Таблиця 6.4
Значення рІ деяких білків
|
Кислі білки |
рІ |
Лужні білки |
рІ |
|
Пепсин |
1,0 |
|
6,4-7,2 |
|
Казеїн |
4, |
Гемоглобін |
7,07 |
|
Желатин |
4,7 |
Гліадін пшениці |
9,8 |
|
Альбумін яйця |
4,8 |
Цітохром С |
10,6 |
В ізоелектричній точці протилежно заряджені групи –NH3+ та –СОО- притягуються одна до одної, і молекула білка закручується у спіраль (рис.6.4, б).
|
|
|
Рис.6.4. Стан білкової молекули при різних значеннях рН: а – рН < рі; б – рн = рі; в – рН > рі. |

При зміщенні рН від ізоелектричної точки в бік зменшення або збільшення рН однойменно заряджені групи відштовхуються, і молекула білка розпрямляється, набуваючи лінійної форми (рис.6.4, а, в).
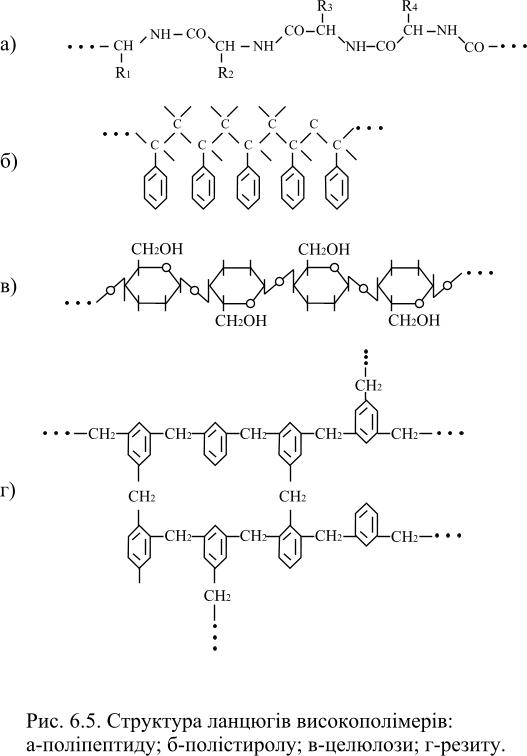
Будова ВМС. Будова молекул високополімерів залежить від будови молекул вихідних мономерів. Маромолекули, що утворюються при полімеризації, можуть мати як лінійну, розгалужену, сітчасту, так і більш складну форму (рис.6.5).
Наприклад, молекули целюлози, крохмалю і глікогену складаються з глюкозних залишків (рис.6.5, в), які з’єднуються між собою через атоми Оксигену. Молекула целюлози має лінійну форму будови, а молекули крохмалю і глікогену складаються з розгалужених ланцюгів, а саме – молекула крохмалю має деревоподібну, а глікогену – кущоподібну форму (рис.6.6).
Це пояснюється тим, що у процесі синтезу лінійне розташування ланок у ланцюгу може порушуватися, при цьому утворюються розгалуження. До утворення просторових або зшитих структур приводить введення природних або спеціальних домішків хімічно активних низькомолекулярних речовин. Наприклад, природний каучук, який характеризується суворо лінійним розташуванням ланок у ланцюгу, при вулканізації (вводиться Сульфур) набуває зшиту структуру.
В макромолекулі полімеру ланки ланцюга сполучені між собою хімічними зв’язками, які навіть при незначному збільшенні відстані між сусідніми атомами Карбону різко послаблюються. Тому під впливом теплового руху взаємне розташування атомів Карбону може змінюватись. Завдяки цьому макромолекула може набувати різноманітних конформацій, тобто гнучкості.
|
|
|
Рис.6.5. Структура ланцюгів високополімерів: а – поліпептиду; б – полістиролу; в – целюлози; г - резиту |
Дуже гнучкі макромолекули прагнуть набути енергетично вигідну сферичну форму і утворювати глобули (рис.6.7, а). Глобули можуть утворюватися з одиничної макромолекули, або з кількох макромолекул (рис.6.7, б, в).
|
|
|
Рис.6.6. Схема зображення макромолекул: а – целюлози; б – крохмалю; в - глікогену |

|
|
|
Рис.6.7. Надмолекулярні структури полімерів: а – макромолекула розтягнута і згорнута в клубок; б- глобула з одиничної макромолекули; в – глобула з кількох макромолекул; г – пачкове утворення; д –макромолекула згорнута у пачку; е – пачка з кількох макромолекул; ж – схема входження макромолекули одночасно в дві пачки; з - фібрила |

Макромолекули можуть складатися у пачки (рис.6.7, г, д, е, ж) утворювати великі і сильно витягнуті агрегати-фібрили (рис.6.7, з). Пластинчасті і голкоподібні структури полімерів можуть утворювати більш складніші і досконаліші структури-сфероліти і навіть монокристали.
Властивості розчинів ВМС. Особливість розчинення ВМС полягає у тому, що розчинність полімерів можлива лише у тих розчинниках, до яких вони ліофільні, тобто гідрофільні полімери розчиняються у воді та інших полярних розчинниках, а гідрофобні полімери – у вуглеводнях. Так, желатина розчиняється у воді, а каучук – у бензені.
Якщо полярність розчинника не співпадає з полярністю ВМС, в цьому випадку при розчиненні утворюються колоїдні розчини, а споріднені ВМС і розчинник утворюють істинні розчини. Наприклад, розчин каніфолі у воді є істинним розчином, а якщо до цього розчину додати спирту – утворюється колоїдний розчин (дисперсія каніфолі у спирті).
Розчиненню
полімеру передує набухання, тобто
проникнення розчинника в порожнині
полімеру. При цьому збільшуються маса
і об’єм зразка полімеру (![]() ).
).
Кінетика
процесу набухання зразка полімеру
приведена на рис.6.8. Кінетичні криві
описують три стадії набухання: початкову
(обмежену) стадію набухання (крива 1),
подальшу стадію набухання з частковим
розчиненням зразка полімеру (крива 2) і
стадію необмеженого набухання з повним
розчиненням зразка полімеру при якому
макромолекули полімеру переходять у
розчин. Ступінь набухання (![]() )
при цьому набуває від’ємного значення
(крива 3).
)
при цьому набуває від’ємного значення
(крива 3).
|
|
|
Рис.6.8. Кінетика набухання полімеру: 1 – обмежене набухання; 2 – набухання з обмеженим розчиненням; 3 – необмежене набухання (повне розчинення) |

Набухання і розчинення ВМС – самовільний процес, що відбувається при зменшенні енергії Гіббса (ΔG<0). На процес впливають такі фактори: в’язкість розчинника, молекулярна маса полімеру, його надмолекулярна структура, температура розчинення. Велике значення має хімічна природа полімеру і розчинника. Так, набуханню і розчиненню сприяє утворення гідрогенних зв’язків між полярними групами полімеру і розчинника.
Підвищення в’язкості розчинника зменшує швидкість проникнення рідини в полімер, а це значно сповільнює процес набухання. Навпаки підвищення температури прискорює процеси набухання і розчинення.
Істотно впливає на процес розчинення зростання молекулярної маси полімеру. Більша довжина макромолекули потребує більшої витрати енергії на розрив усіх зв’язків в макромолекулі, тому розчинність зменшується.
Надмолекулярна структура високополімерів також значно впливає на процеси набухання і розчинення ВМС. Так, у щільно згорнуті полімерні клубки альбуміну білків, глікогену молекули розчинника дуже важко проникають, тому такі глобулярні полімери практично не набухають.
В той же час, якщо глобули полімерів мають велику спорідненість до розчинника, їх взаємодія послаблює зв’язки між окремими глобулами і вони переходять у розчин.
Гірше і важче набухають і розчиняються полімери з розгалуженими мікромолекулами і кристалічні полімери. Перші мають між макромолекулами поперечні зшивки, які значно зменшують проникнення розчинника і повністю виключають можливість розчинення полімеру. Другі взаємодіють з розчинником лише поверхнею контакту, що обмежує набухання і розчинення кристалічного полімеру.
|
|
|
Рис.6.9. Вплив рН на ступінь набухання білків |

Набухання поліелектролітів у воді в значній мірі залежить від стану окремих ланок молекули білка. В ізоелектричній точці білка (рІ) протилежно заряджені групи –NH3+ та –СОО- притягуються і молекула білка закручується у спіраль (згортається). Проникнення розчинника у макромолекулу зменшується, тому в ізоелектричній точці набухання мінімальне. При зміні рН в кислий або лужний бік від ізоелектричної точки однойменно заряджені групи відштовхуються, і молекула білка розпрямляється, що призводить до зростання набухання. Залежність ступеню набухання білків від рН приведена на рис.6.9.
Добування і властивості дисперсних систем і розчинів ВМС. Виникнення дисперсної системи в результаті утворення частинок нової стабільної фази в метастабільної системі може бути викликана як відхиленням в хімічному складі фаз, так і за рахунок фізико-хімічного впливу на систему. Це в першу чергу різні хімічні реакції, які приводять до виникнення високих концентрацій малорозчинних сполук, що обумовлює пересичення в системі, а також зміна тиску, температури і складу фаз.
Процеси утворення дисперсних систем різної дисперсності і концентрації широко розповсюджені в природі і використовуються в різних областях технології. Так, окисно-відновні реакції лежать в основі багатьох методів добування золів, наприклад,
![]()
![]()
![]()
Важливе значення мають утворення гідрозолей в процесах гідролізу солей.
![]()
хімічна, або дисолюційна пептизація:
Fe(OH)3 + HCl L FeOCl + 2H2O,
обмінні реакції:
![]()
які можуть застосовуватися для добування дисперсних систем.
Метод пептизації полягає у дезагрегації частинок осаду розпушеної структури, між якими є прошарок дисперсійного середовища. За механізмом пептизація поділяється на дисолюційну, адсорбційну і пептизацію промиванням розчинника (дисперсійним середовищем).
Адсорбційна пептизація – це вибіркова адсорбція на осаді йонів, електролітів, що додаються до осадів.
При хімічній, або дисолюційної пептизації речовини, що додаються до осадів, вступають в реакцію з поверхнею осадів.
Промивання осадів розчинником або дисперсійним середовищем, створює вимивання надлишку електроліту і утворюється стійкий золь. Пептизація може відбуватися при додаванні до осаду неелектролітів, молекули яких адсорбуються на частинках осаду і запобігають їх злипанню. Такими речовинами є органічні кислоти, багатоатомні спирти, тощо.
Для одержання колоїдних розчинів велике практичне значення мають методи диспергування, що грунтуються на подрібненні великих частинок до колоїдного стану. Процеси диспергування потребують великих енергетичних витрат. Для добування високодисперсних систем використовують колоїдні млини.
Важливу роль в інтенсифікації процесів диспергування грає введення поверхнево-активних середовищ, що значно зменшує енергетичні витрати на ці процеси.
Високодисперсні золі металів і сплавів у самих різних дисперсійних середовищах можуть бути одержані методом електророзпорушення, механічним або акустичним диспергуванням.
Електричні явища. Завдяки надлишку поверхневої енергії, якою володіють дисперговані часточки, на межі розділу фаз на міжфазових поверхнях виникає взаємодія, що приводить до утворення подвійного електричного шару (ПЕШ).
Просторове розділення зарядів і утворення ПЕШ характерно для любої межі двох фаз, в системі якої є йони і інші заряджені частинки. Причиною утворення ПЕШ і відповідного стрибка потенціалу можуть бути:
а) обмін зарядженими частинками;
б) вибіркова адсорбція йонів;
в) адсорбція полярних молекул.
Знак заряду поверхні частинок залежить від природи дисперсної фази та дисперсійного середовища. При змочуванні водою поверхня таких речовин, як метали, сульфіди металів, силікагель, крохмаль, деревина, папір, гуміарабік тощо заряджається негативно, а нерозчинних оксидів, гідроксидів металів, деяких солей – позитивно.
На знак заряду поверхні частинок значно впливає значення діелектричної проникності. З двох спряжених фаз позитивно заряджається фаза з більшою діелектричною проникністю, наприклад, частинки глини у воді заряджені негативно, а вода – позитивно.
Подвійний електричний шар складається з двох частин: щільної (внутрішня обкладка) і дифузної (зовнішня обкладка( (рис.6.10). Більш близька до поверхні внутрішня обкладка ПЕШ (шар Штерна-Гельмгольца) відповідає частині, де адсорбційні сили суттєві. У більш віддаленій дифузній частині (шар Гуї-Чепмена) ними можна знехтувати.
|
|
|
Рис.6.10. Схематична будова подвійного електричного шару: 1 – внутрішня обкладка (адсорбційний шар); 2 – зовнішня обкладка (дифузний шар); 3 – молекули дисперсійного середовища |

Внутрішня обкладинка представляє собою адсорбційний монойонний шар, товщиною не менше двох радіусів йонів (б) (рис.6.11). За Штерном в неї знаходяться потенціаловизначаючі йони, фіксовані твердою поверхнею і частина протийонів адсорбційного шару.
Зовнішня обкладинка складається з протийонів дифузного шару. Її товщина (λ) може бути значною і залежить від властивостей і складу дисперсної системи. Розрахувати її можна за рівнянням:
![]() ,
(6.6)
,
(6.6)
де
![]() - діелектрична проникність дисперсійного
середовища;
- діелектрична проникність дисперсійного
середовища;
![]() - діелектрична стала; І
– йонна сила розчину; F
– число Фарадея.
- діелектрична стала; І
– йонна сила розчину; F
– число Фарадея.
На
межі твердої та рідкої фаз виникає так
званий поверхневий, або
![]() -потенціал.
Величина
-потенціал.
Величина
![]() - потенціалу пропорційна числу заряджених
йонів твердої поверхні (ядра), а його
знак збігається зі знаком потенціаловизначаючих
йонів.
- потенціалу пропорційна числу заряджених
йонів твердої поверхні (ядра), а його
знак збігається зі знаком потенціаловизначаючих
йонів.
На
межі адсорбційного і дифузного шарів
(лінія АА)
виникає стрибок потенціалу, який Штерн
назвав
![]() - потенціалом, або адсорбційним
потенціалом. Потенціал у адсорбційному
шарі зменшується лінійно (пряма MN)
від
- потенціалом, або адсорбційним
потенціалом. Потенціал у адсорбційному
шарі зменшується лінійно (пряма MN)
від
![]() до
до
![]() .
.
|
|
|
Рис.6.11.
Схема будови подвійного електричного
шару за Штерном: |

Заряд адсорбційного шару складається із зарядів йонів, що адсорбуються як за рахунок електростатичного адсорбційного потенціалу, так і за рахунок потенціалу специфічної адсорбції.
У
дифузному шарі протийони розподілені
нерівномірно, тому потенціал у дифузній
частині ПЕШ змінюється за експоненціальним
законом Больцмана (крива NK)
від значення
![]() наближуючись до нуля:
наближуючись до нуля:
![]() ,
(6.7)
,
(6.7)
де
-
![]() - ефективна товщина дифузного шару
(відстань на який
- ефективна товщина дифузного шару
(відстань на який
![]() - потенціал зменшується в е
разів); х
– відстань від межі шару Штерна в глибину
рідкої фази.
- потенціал зменшується в е
разів); х
– відстань від межі шару Штерна в глибину
рідкої фази.
Товщина ПЕШ залежить від йонної сили розчину (І):
![]() .
(6.8)
.
(6.8)
Згідно теорії сильних електролітів Дебая-Гюккеля з підвищенням концентрації електролітів у рідині і зарядоутворюючих їх йонів товщина ПЕШ зменшується.
Важливим
потенціалом, який характеризує ПЕШ є
електрокінетичний потенціал, або
![]() (дзета)-потенціал.
(дзета)-потенціал.
![]() -потенціал
виникає на межі сковзання (лінія ВВ).
Межа сковзання – це межа між часточками,
здатними до руху в електричному полі,
та оточуючою її рідиною. Електрокінетичний
потенціал розраховують за експериментальними
даними за рівнянням Гельмгольца-Смолуховського:
-потенціал
виникає на межі сковзання (лінія ВВ).
Межа сковзання – це межа між часточками,
здатними до руху в електричному полі,
та оточуючою її рідиною. Електрокінетичний
потенціал розраховують за експериментальними
даними за рівнянням Гельмгольца-Смолуховського:
![]() ,
(6.9)
,
(6.9)
де
η
- в'язкість середовища;
![]() - об’ємна швидкість руху середовища;
- об’ємна швидкість руху середовища;
![]() -
питома електропровідність;
-
питома електропровідність;
![]() -
діелектрична стала (8,854 .
10-12
ф/м);
-
діелектрична стала (8,854 .
10-12
ф/м);
![]() - діелектрична проникність середовища;
І –
сила струму.
- діелектрична проникність середовища;
І –
сила струму.
|
|
|
Рис.6.12. Схема, що ілюструє термодинаміку потенціалів |

З
точки зору термодинаміки
![]() -
потенціал дорівнює роботі переносу
одиниці позитивного заряду з глибини
даної фази в точку, що розташована у
безпосередній близкості від поверхні
даної фази (10-6
м) (рис.6.12).
-
потенціал дорівнює роботі переносу
одиниці позитивного заряду з глибини
даної фази в точку, що розташована у
безпосередній близкості від поверхні
даної фази (10-6
м) (рис.6.12).
Термодинамічно
![]() -потенціал
відповідає роботі, яка відповідає
переносу одиничного заряду з нескінченно
віддаленої точки об’єму рідкої фази з
потенціалом, рівним нулю, на межу
сковзання з потенціалом
-потенціал
відповідає роботі, яка відповідає
переносу одиничного заряду з нескінченно
віддаленої точки об’єму рідкої фази з
потенціалом, рівним нулю, на межу
сковзання з потенціалом
![]() .
.
Величина
електрокінетичного потенціалу залежить
від концентрації електролітів, що
присутні у розчині, а також від валентності
йонів, особливо, протийонів. Збільшення
концентрації йонів у розчині призводить
до стискання дифузного шару до товщини
![]() і зменшенню
і зменшенню
![]() -потенціалу
до нуля. В цьому випадку дисперсна
система переходить в ізоелектричний
стан.
-потенціалу
до нуля. В цьому випадку дисперсна
система переходить в ізоелектричний
стан.
Особливо
великий вплив на
![]() -потенціал
виявляють органічні катіони, гідрогенйони,
ряд деяких йонів, які не тільки здатні
дуже знизити величину електрокінетичного
потенціалу, але й викликати зміну його
знаку. Це явище називають перезарядкою.
На рис.6.13 зображена схема , яка пояснює
зміну знаку
-потенціал
виявляють органічні катіони, гідрогенйони,
ряд деяких йонів, які не тільки здатні
дуже знизити величину електрокінетичного
потенціалу, але й викликати зміну його
знаку. Це явище називають перезарядкою.
На рис.6.13 зображена схема , яка пояснює
зміну знаку
![]() -потенціалу.
Крива 1 зміни потенціалу до перезарядки
відсікає на межі сковзання відрізок,
який відповідає величині позитивного
-потенціалу.
Крива 1 зміни потенціалу до перезарядки
відсікає на межі сковзання відрізок,
який відповідає величині позитивного
![]() -потенціалу.
Крива 2 падіння потенціалу відсікає на
межі сковзання відрізок, який розташований
під віссю абсцис, що вказує на надлишок
в адсорбційному шарі протийонів. Це
відповідає негативному значенню
-потенціалу.
Крива 2 падіння потенціалу відсікає на
межі сковзання відрізок, який розташований
під віссю абсцис, що вказує на надлишок
в адсорбційному шарі протийонів. Це
відповідає негативному значенню
![]() -потенціалу.
-потенціалу.
|
|
|
Рис.6.13.
Схема зміни знаку
до перезарядки; 2 – крива падіння потенціалу |

Зниження концентрації колоїдного
розчину при розбавленні призводить до
десорбції потенціаловизначаючих йонів,
що зменшує значення поверхневого
![]() -
потенціалу. При розведенні колоїдного
розчину значення
-
потенціалу. При розведенні колоїдного
розчину значення
![]() -
потенціалу збільшується, але зменшення
-
потенціалу збільшується, але зменшення
![]() -потенціалу
призводить і до зменшення
-потенціалу
призводить і до зменшення
![]() -потенціалу.
Аналогічно впливає підвищення температури:
збільшується товщина дифузного шару,
але одночасно зменшується
-потенціалу.
Аналогічно впливає підвищення температури:
збільшується товщина дифузного шару,
але одночасно зменшується
![]() -
потенціал зі зростанням десорбції
потенціаловизначаючих йонів.
-
потенціал зі зростанням десорбції
потенціаловизначаючих йонів.
Будова міцел. Розглянемо будову колоїдних часточок ліофобних золів на прикладі золю силікатної кислоти. Згідно з міцелярною теорією будови колоїдних розчинів золь складається з міцел та інтерміцелярної рідини (дисперсійного середовища). Міцела – це часточка дисперсної фази разом з зарядами, які утворюють ПЕШ. Зарядами у даному випадку є йони, які утворюються під час дисоціації і гідролізу речовин:
Na2CO3 L 2Na+ + CO32-,
HCl L H+ + Cl-,
CO32- + HOH L HCO3- + OH-.
В середині міцели утворюється агрегат, який складається з m молекул H2SiO3. Згідно з правилом Панета-Фаянса, йони SiO32- адсорбуються на поверхні агрегату, утворюючи шар потенціалвизначаючих йонів, які, в свою чергу, наближують (притягають) до себе частину протийонів х . Н+. Агрегат разом з адсорбованими на ньому потенціаловизначаючими йонами називається ядром міцели. Ядро і адсорбований шар протийонів складають колоїдну часточку або гранулу. Колоїдна часточка має величину заряду, який складається з суми потенціловизначаючих йонів і протийонів адсорбційного шару. Знак заряду колоїдної часточки завжди такий самий, який має потенціаловизначаючий йон. Заряд гранули компенсується зарядами йонів протилежного знаку дифузного шару, тому в цілому міцела є електронейтральною часточкою. Отже, у міцели розрізняють агрегат, ядро, адсорбційний шар, колоїдну часточку і дифузний шар протийонів:
![]()
агрегат адсорбційний протийони
шар дифузного
ядро шару
колоїдна часточка (гранула)
Формула міцели може мати і інший вигляд:
![]()
Якщо в якості потенціаловизначаючих йонів будуть йони HSiO3-, формула міцели буде мати вигляд:
![]()
Залежно від того, який з йонів в розчині буде мати більшу концентрацію, будова міцели змінюється. Так, при надлишку в розчині йонів Н+, міцела набуває вигляду:
![]()
Колоїдні часточки золю силікатної кислоти при агрегації переходять у драглі. Висушені драглі представляють собою часточки, які покриті групами ≡SiOH. Формула міцели таких драглів (силікагелю) має вигляд:
![]()
де, nSiO- означає не адсорбовані йони (таких йонів взагалі не існує), а число дисоційованих груп ≡SiOH (рис.6.14).
|
|
|
Рис.6.14. Схема будови гідратованого оксиду силіцію(IV) |

Формули міцел золів Ауруму, Аргентуму, Феруму за реакціями, при яких вони утворюються, мають вигляд:
![]()
![]()
![]()
![]()
Утворення золю Феруму за методом пептизації промивання розчинником відбувається за схемою:
{[m[Fe(OH)3] . nFe3+]3n+ . 3nCl-}0 L
L {[m[Fe(OH)3] . nFe3+]3+ . 3(n-x)Cl-}3x+ . 3xCl-.
Електрокінетичні явища. Особливі електричні властивості дисперсних систем вперше виявив російський вчений Ф.Рейсс в 1807 р. Встановивши у шматок мокрої глини дві скляні трубки, насипавши потроху піску і заливши трубки чистою водою, він занурив у воду електроди джерела постійного електричного струму (рис.6.15.). При пропусканні електричного струму крізь цю систему Рейсс помітив перенос рідини з анодного в катодний простір, а часточки глини проникали в анодний простір; таке явище отримало назву електрофорезу, або катафорезу.
|
|
|
Рис.6.15. Схема приладу Рейсса |

Механізм електрофорезу полягає в тому, що під дією електричного поля ПЕШ йонів розривається на межі сковзання, часточки глини, які мають негативний заряд рухаються до анода, а позитивно заряджені молекули води в протилежний бік до катода (рис.6.16). Явища взаємного переміщення твердої і рідкої фаз під впливом електричного струму називаються електрокінетичними.
|
|
|
Рис.6.16. Схема руху часточок при електрофорезі |

Швидкість
електрофорезу залежить від величини
![]() -потенціалу
і визначається рівнянням
Гельмгольца-Смолуховського:
-потенціалу
і визначається рівнянням
Гельмгольца-Смолуховського:
![]() (6.10)
(6.10)
де Е – зовнішня різниця потенціалів; l – віддаль між електродами; U0 – лінійна швидкість руху фаз.
В рівнянні відношення Е/l є градієнтом потенціалу Н, тобто напругою зовнішнього електричного поля:
![]()
Лінійна швидкість руху часточок U0 змінюється пропорційною напругою зовнішнього електричного поля Н, тому не може служити їх характеристикою. При одиничному градієнті потенціалу (H=1)
![]() (6.11)
(6.11)
де UЕФ – електрофоретична рухомість.
Електрофоретичну рухомість, яка є характеристикою руху колоїдних часточок, знаходять експериментально за виразом:
![]() (6.12)
(6.12)
де
h
– шлях пройдений часточкою за час
![]() ;
l
– відстань між електродами; Е
– прикладена різниця потенціалів.
;
l
– відстань між електродами; Е
– прикладена різниця потенціалів.
|
|
|
Рис.6.17. Схема досліду Рейсса з електроосмосу |

В іншому досліді Рейсс дослідив переміщення рідини крізь порувату перегородку з товченого кварцу (рис.6.17) при пропусканні сталого струму. Рівень води в трубці з позитивним електродом знижувався, а з негативним – підвищувався. Рух рідини в трубках відбувався доти, поки не встановлювалась певна різниця рівнів рідини (h – гідростатичний тиск). З цього досліду Рейсс зробив висновок, що при контакті з частинками кварцу рідина заряджається позитивно і це є причиною підняття рівня води в трубці з від’ємно зарядженим електродом. Це явище спрямованого переміщення дисперсійного середовища відносно нерухомої дисперсної фази в постійному електричному полі одержало назву електроосмосу.
Напрямок
переміщення рідини (рис.6.18) вимірювання
швидкості її течії дає можливість
визначити знак
![]() -потенціалу
і розрахувати стрибок потенціалу на
межі сковзання.
-потенціалу
і розрахувати стрибок потенціалу на
межі сковзання.
|
|
|
Рис.6.18. Схема руху рідини в капілярі при електроосмосі |

Швидкість руху дисперсійного середовища, віднесену до одиниці напруженості електричного поля, називають електроосмотичною рухомістю (UЕОР). Її визначають, як:
![]() (6.13)
(6.13)
де
U0
– лінійна швидкість течії рідини; v
– об’ємна швидкість течії рідини
(v=VР/![]() - об’єм переміщеної рідини (VР)
за час (
- об’єм переміщеної рідини (VР)
за час (![]() ));
));
![]() -
питома електропровідність; І
– сила струму.
-
питома електропровідність; І
– сила струму.
В
цьому випадку величину
![]() -потенціалу
обчислюють за рівнянням:
-потенціалу
обчислюють за рівнянням:
![]() (6.14)
(6.14)
Явище, протилежне електроосмосу, є потенціал течії або перебігу (Квінке, 1859 р.). Потенціал, який виникає при протіканні рідини під тиском через пористу мембрану, називають потенціалом протікання. Він виникає внаслідок руху зарядів (йонів дифузного шару) вздовж межі поділу фаз в напрямку потоку рідини. Ця різниця потенціалів на кінцях капілярів діафрагми приводить до появи струму провідності в зворотному напрямку. При рівновазі струмів потенціал протікання (UПРОТ) дорівнює:
![]() (6.15)
(6.15)
де
![]() Р
– тиск, який викликає течію рідини.
Р
– тиск, який викликає течію рідини.
При осіданні часточок дисперсної фази по висоті стовпа рідини виникає різниця потенціалів (ефект Дорна, 1878р.). Це явище обернене електрофорезу. Потенціал, який виникає, називається потенціалом седиментації. Причиною цього явища є розділення йонів дифузних шарів і колоїдних часточок, що рухаються при осіданні, внаслідок їх тертя об шар рідини. При цьому дифузні йони відстають від колоїдних часточок і по висоті виникає різниця потенціалів.
|
|
|
Рис.6.19. Схема експерименту Дорна з седиментації |

Величину потенціалу седиментації (UСЕД) обчислюють за рівнянням:
![]() (6.16)
(6.16)
де
![]() - об’ємна доля дисперсної фази (для
сферичних часточок радіусом r і
кількістю n
- об’ємна доля дисперсної фази (для
сферичних часточок радіусом r і
кількістю n
![]() дорівнює
дорівнює
![]() ;
ρ і ρ0 - густина дисперсної
фази і дисперсійного середовища; q
– прискорення сили тяжіння.
;
ρ і ρ0 - густина дисперсної
фази і дисперсійного середовища; q
– прискорення сили тяжіння.
Значення
![]() -потенціалу
часточок колоїдних розчинів знаходиться
в межах 1,5-75 мВ. Електрофоретична
рухливість часточок золів має значення
порядку 0,4-0,8.10-8 м-2/с.В,
а експериментальні значення
електрофоретичної рухливості часточок
досягають
-потенціалу
часточок колоїдних розчинів знаходиться
в межах 1,5-75 мВ. Електрофоретична
рухливість часточок золів має значення
порядку 0,4-0,8.10-8 м-2/с.В,
а експериментальні значення
електрофоретичної рухливості часточок
досягають
![]() 5,0
. 10-8 м-2/c.В.
5,0
. 10-8 м-2/c.В.
Методи електрофорезу дають змогу аналізувати суміші біологічних рідин, розділяти їх на компоненти, визначити чистоту біологічних препаратів. Електрофорез застосовують в медичній діагностиці, в лікувальній справі, при нанесенні оксидів металів на поверхні радіовиробів тощо.
Методи електроосмосу мають велике практичне застосування для концентрування колоїдних систем, зневоднення пористих матеріалів, осушення грунтів, будівельних конструкцій.
Виникнення потенціалів протікання і седиментації є причинами пожеж і вибухів танкерів, нафтосховищ, нафтопроводів, технологічних комунікацій, виникнення блискавок і грозових розрядів.
Оптичні властивості дисперсних систем і розчинів ВМС. При розповсюдженні світла в деякому середовищі відбуваються такі явища, як заломлення, поглинання, відбиття і розсіювання світла. Оптичні явища надзвичайно важливі для вивчення дисперсних систем. Їх використання дає змогу досліджувати будову і властивості колоїдних часточок і їх угрупувань, визначати концентрацію, вплив різних факторів на взаємодію променів світла з часточками дисперсних систем.
Розсіювання світла характерно для дисперсних систем, коли лінійні розміри часточок менші за довжину хвилі падаючого світла. В цьому випадку спостерігається дифракція – світлові хвилі огинають часточки і змінюють свій початковий напрям руху. Внаслідок дифракції в мікронеоднорідному середовищі дисперсної системи відбувається матове (блакитне) світіння (опалесценція). Цим пояснюється явище – конус Тіндаля (ефект Фарадея-Тіндаля) при якому промінь світла в дисперсній системі стає видимим.
Найбільш простішим випадком є розсіювання світла при виконанні наступних умов:
а) найбільший розмір часточок (r) значно менше довжини хвилі падаючого світла (λ), тобто:
η = λ /10;
б) часточки не поглинають світло (не забарвлені);
в) часточки оптично ізотропні;
г) концентрація часточок мала – відстань між часточками велика порівняно з довжиною хвилі падаючого світла;
д) часточки не проводять електричний струм;
е) об’єм дисперсної системи дуже малий, що дозволяє нехтувати вторинним розсіюванням розсіяного світла.
За Д.Релеем (1871р.), якщо на часточку падає поляризоване світло, то загальна кількість світлової енергії, що розсіюється одиницею об’єму дисперсної системи (ІР) дорівнює:
![]() (6.17)
(6.17)
де

І0
– інтенсивність падаючого світла;
![]() -
число часточок в одиниці об’єму; V
– об’єм однієї часточки; λ - довжина
хвилі падаючого світла; n1
і n2 – показники заломлення
дисперсної фази і дисперсійного
середовища.
-
число часточок в одиниці об’єму; V
– об’єм однієї часточки; λ - довжина
хвилі падаючого світла; n1
і n2 – показники заломлення
дисперсної фази і дисперсійного
середовища.
За допомогою рівняння Релея визначають об’єм, радіус і концентрацію часточок дисперсної системи.
З наведеного рівняння Релея випливає, що якщо n1 = n2, світло системою не розсіюється.
За стаціонарних умов вимірювання величини n1, n0 і λ є сталими, тоді рівняння (6.17) спрощується:
![]() (6.18)
(6.18)
Рівняння Релея застосовується тільки для систем, розмір часточок яких не більше 0,1 частини довжини світлової хвилі. Якщо часточки мають більші розміри, то величина світлорозсіювання змінюється обернено пропорційно вже не в четвертій, а меншій степені довжини хвилі світла. Це сприяє світлорозсіюванню. При розмірах часточок, що значно перевищують довжину світлової хвилі, світлорозсіювання переходить у відбиття, яке не залежить від довжини хвилі.
При проходженні променя білого світла через дисперсну систему в більшій мірі розсіюються більш короткі хвилі, тобто синьо-фіолетової частини спектру. Так, розсіювання найбільш короткохвильової частини сонячного світла на флуктуаціях густини земної атмосфери надає небу вдень голубого кольору, а при сході і заході сонця – червоного. Це стає тому, що їх спостереження відбувається через нижні шари атмосфери, що містять значного розміру часточки пилу і диму. Цей ефект особливо підсилюється при вітряній погоді, коли концентрація часточок збільшується.
Зменшення інтенсивності світла dI при проходженні крізь шар чистої речовини, згідно із законом Ламберта, пропорційне інтенсивності світла І і товщині шару dl:
![]() (6.19)
(6.19)
або після перетворень
![]() або ІПР = І0
. е-kl,
(6.20)
або ІПР = І0
. е-kl,
(6.20)
де І0
і ІПР – інтенсивність
падаючого світла і яке пройшло крізь
систему; k – коефіцієнт екстинкції (![]() ,
м-1); 1е – товщина
шару речовини, що ослаблює інтенсивність
світла в е разів – середня глибина
проникнення світла.
,
м-1); 1е – товщина
шару речовини, що ослаблює інтенсивність
світла в е разів – середня глибина
проникнення світла.
У випадку розчинів речовин світлопоглинання збільшується зі збільшенням концентрації (закон Ламберта-Бера):
![]() або ІПР = І0
. е-kl,
(6.21)
або ІПР = І0
. е-kl,
(6.21)
де с – концентрація речовини.
Закон Ламберта-Бера дійсний тільки для відносно тонких шарів розбавлених розчинів.
Сумарне розсіювання розчинами високомолекулярних сполук при освітленні системи неполяризованим світлом описується рівнянням Релея у вигляді:
![]() (6.22)
(6.22)
де

Відношення ІР/I0
називається каламутністю системи
![]() ,
яку визначають за виміряними значеннями
оптичної густини D:
,
яку визначають за виміряними значеннями
оптичної густини D:
![]() (6.23)
(6.23)
де D = lg I0/IПР; 1 – товщина шару розчину, крізь який проходить світло.
Для розчинів ВМС з часточками, розмір яких перебуває в межах 0,1λ<d<0,3λ рівняння (6.22) треба записати так:
![]() або
(6.24)
або
(6.24)
![]() (6.25)
(6.25)
де х – величина, яка є функцією розміру часточок.
Величину х визначають як тангенс кута нахилу прямої в координатах рівняння (6.24) або (6.25) у логарифмічній формі:
![]() (6.26)
(6.26)
![]() (6.27)
(6.27)
За знайденою величиною х визначають розміри часточок.
Розмір часточок можна визначити спостерігаючи їх в ультрамікроскопі при бічному освітленні. В геометричному об’ємі:
![]()
де 1 – сторона квадрата, яку визначають за допомогою окуляра-мікрометра, розчин з масовою концентрацією дисперсної фази С і кількістю часточок в об'ємі n при густині речовини дисперсної фази ρ має розмір часточок d, який розраховується за рівнянням:
![]() (6.28)
(6.28)
Явище розсіювання світла використовують в оптичних методах вивчення колоїдних систем, визначення концентрації золів, розмірів колоїдних часточок, з метою світломаскування, світлової сигналізації, визначення молекулярних мас високомолекулярних сполук, тощо.
Стійкість дисперсних систем та розчинів ВМС. Ліофобні, тобто термодинамічно нерівноважні дисперсні системи мають нахил до процесів, що ведуть до зміни їх будови – дисперсності, характеру розподілу часточок за розміром в об’ємі дисперсійного середовища. За поглядами М.Пескова розрізняють седиментаційну і агрегативну стійкість дисперсних систем.
Седиментаційна стійкість – це стійкість системи проти зниження потенціальної енергії часточок дисперсної фази при їх осіданні під дією сил тяжіння.
Агрегативна стійкість – це здатність системи протидіяти процесам, які ведуть до зменшення вільної енергії поверхні поділу часточок дисперсної фази з дисперсійним середовищем.
Процесами руйнування дисперсних систем є ізотермічна перегонка (кристалізація) речовини від малих часточок до більш крупних, коалесценція (зливання часточок) і коагуляція (агрегування часточок при їх злипанні).
Роль процесів ізотермічної перегонки, коалесценції і коагуляції в порушенні агрегативної стійкості дисперсних систем різна, поперед всього, в залежності від фазового стану дисперсійного середовища. Коалесценція, коагуляція і седиментаційний розподіл властиві системам з легкорухомим (рідким або газоподібним) дисперсійним середовищем.
Природа стійкості дисперсних систем і умови протікання різних процесів їх руйнування суттєво залежать від концентрації дисперсної фази, характеру взаємодії часточок одна з одною і т.д. Найбільш нестійкими є гідрофобні колоїдні системи, для яких характерна слабка взаємодія між часточками дисперсної фази і дисперсійного середовища. Вони на відміну від гомогенних систем мають великий запас вільної поверхневої енергії, зменшення якої відбувається внаслідок зменшення поверхні поділу фаз. Це призводить до злипання часточок, тому коагуляція є термодинамічно вигідним і довільним процесом. Стабілізація таких систем обумовлена наданням певних факторів стійкості.
У реальних умовах в дисперсних системах одночасно діють декілька факторів. Це термодинамічні (ентропійний, електростатичний, адсорбційно-сольвативний) та кінетичні (гідродинамічний, структуро-механічний). Домінуючими з них є два фактори: адсорбційно-сольвативний та електростатичний.
Адсорбційно-сольвативні процеси сприяють зменшенню міжфазового поверхневого натягу та енергії Гіббса поверхні поділу фаз.
Електростатичний фактор обумовлений
створенням електростатичних сил
відштовхування, які зростають зі
збільшенням потенціалу поверхні часточок
![]() і особливо електрокінетичного
і особливо електрокінетичного
![]() -потенціалу.
-потенціалу.
Іншим фактором стабілізації є процеси ліофілізації. Самовільне утворення ліофільних колоїдних систем обумовлено приростом вільної поверхневої енергії, який при диспергуванні макрофаги компенсується виграшем вільної енергії внаслідок збільшення ентропії за рахунок включення відокремлених часточок у броунівськи рух. Так, наприклад, природні глини при змочуванні водою внаслідок інтенсивної сольватації розпадаються на окремі часточки і утворюють агрегативно стійкі системи.
Ліофілізація особливо характерна для систем, які містять міцелоутворюючі ПАР та ВМС. При утворенні цих систем вільна енергія Гіббса зменшується (ΔG<0).
Агрегативна і седиментаційна стійкості дисперсних систем взаємопов’язані. Доки колоїдна система зберігає агрегативну стійкість, вона стійка і седиментаційно. Порушення агрегативної стійкості призводить до коагуляції системи. З часом, якщо в системі при агрегуванні утворюються досить великі угрупування, то система втрачає седиментаційну стійкість, відбувається розділення фаз дисперсної системи і утворюється осад.
Утворення під час коагуляції крупних агрегатів не завжди призводить до утворення осаду. Причиною цього є так звана конденсаційна (фазова) стійкість дисперсних систем, яка пов’язана з структурою та міцністю агрегатів, що утворюються під час коагуляції системи.
За фазовою стійскістю дисперсійні системи поділяються на конденсаційно стійкі і конденсаційно нестійкі системи.
Конденсаційно стійкі системи утворюють при коагуляції нестійкі агрегати, які перетворюються в пухкий осад. Частинки такого осаду втрачають свою індивідуальність і рухомість, але, внаслідок наявності між агрегатами тонких прошарків рідини, зберігаються як окремі утворення досить довгий час. При певних умовах такі агрегати можуть знову розпадатись на окремі часточки, тобто відбувається їх пептизація.
В конденсаційно нестійких системах утворення агрегатів характеризуються міцною структурою, яка обумовлена наявністю в них безпосередніх фазових контактів у вигляді кристалізаційних містків, зрощування частинок. Такі структури є необоротними і утворюють щільні, кристалічні осади.
Коагуляція дисперсних систем може бути зумовлена різними зовнішніми впливами: додаванням електролітів, струшенням, механічним перемішуванням, підвищенням температури або заморожуванням, дією ультразвукового поля, йонізуючих випромінювань, електролізом тощо.
Найбільш ефективним фактором, що викликає коагуляцію гідрофобних золів є дія електролітів. Вони дуже швидко і різко нейтралізують і стискують подвійний електричний шар міцел, що порушує агрегативну стійкість системи.
Коагуляція гідрофобних золів здійснюється постадійно. На першій стадії коагуляції, яку називають прихованою, часточки дисперсної фази наближуються одна до іншої і взаємно фіксуються (зчеплюються), утворюючі агрегати з декількох часточок між якими містяться прошарки дисперсійного середовища (рис.6.20).
|
|
|
Рис.6.20. Стадії коагуляції золів: 1 – стійка дисперсна система; 2 – прихована коагуляція; 3- наявна коагуляція (седиментація) |

На другій стадії коагуляції, яку називають наявною, відбувається більш глибокий процес: прошарки середовища в агрегаті стискуються і між часточками утворюється безпосередній контакт. Структура таких агрегатів стає міцною внаслідок кристалізації і зрощування часточок. Тверді агрегати під дією гравітаційних сил тяжіння осідають утворюючі осад. У системах з рідкою чи газоподібною дисперсною системою на цій стадії відбувається повне злиття часточок (коалесценція).
Коагуляція під дією електролітів. Досліджуючи коагуляцію Шульце і Гарді встановили (1882-1900 р.р.), що коагуляцію викликає не вся молекула електроліту, а лише йон–коагулятор. Йон-коагулятор завжди має заряд протилежний заряду колоїдної гранули.
Згідно з правилом Шульце-Гарді (правило валентності), коагулююча дія йона-коагулятора зростає зі зростанням заряду йона.
Мінімальна концентрація електроліту, що зумовлює повну коагуляцію колоїдної системи називається порогом коагуляції (ПК) і виражається в ммоль/л. Поріг коагуляції обернено пропорційний заряду коагулюючих йонів у шостому степені (закон шостого степеня Дерягіна):
![]() (6.29)
(6.29)
де z – заряд йона, що зумовлює коагуляцію.
Критична концентрація йона-коагулятора СК для тривалентного і двовалентного йонів у сотні і в десятки разів менша ніж для одновалентного:
![]()
Величину, обернену порогу коагуляції, називають коагулюючою здатністю (об’єм колоїдної системи, що скоагульована 1ммолем йона-коагулятора) і позначають VК:
![]() (6.30)
(6.30)
Підвищена коагулюючи здатність йона-когулятора зі збільшенням його заряду пояснюється більш сильним ефектом стиснення подвійного електричного шару колоїдної часточки.
Ефект стиснення ПЕШ у ряда йонів з однаковим зарядом залежить від радіуса сольватованого йона:
Li+ < Na+ < K+ < Rb+ < Cs+; Mg2+ < Ca2+ < Cr2+ <Ba2+;
Cl- < Br - < NO3- < J- < CNS -.
Їх коагулююча активність збільшується зі зменшенням радіуса. Це стосується неорганічних сполук. У випадку органічних йонів їх коагулююча дія зростає зі збільшенням адсорбційної здатності.
Згідно з правилом Траубе для органічних електролітів, які містять карбонвмісні ланцюги різної довжини, коагулюючи здатність рівномірно збільшується зі зростанням СН2-груп.
|
|
|
Рис.6.21. Схема утворення тонкої плівки рідини (2) при наближенні двох об’ємів фази (1) |

Теорія стійкості і коагуляції дисперсних систем. Сучасну теорію стійкості колоїдних систем розробили Б.Дерюгін, Л.Ландау, Е.Вервей, Я.Овербек (1937-1991 р.р.), яку називають теорією ДЛФО. Згідно з теорією ДЛФО при наближенні двох сферичних часточок однакового радіуса r на відстань між їх центрами R у мінімальній ширині щілини h=R-2r виникає розклинювальний тиск рідини II (рис.6.21, а).
Розклинювальний тиск розглядають як надлишковий (порівняно з об’ємами фаз 1 і 2) (рис.6.21, б) тиск, який діє з боку прошарку на поверхні, що обмежують його, і намагається розсунути („розклинити”) їх. Цей тиск визначається молекулярними силами притягання і електростатичними силами відштовхування.
Для визначення електростатичної
складової розклинювального тиску
розглянемо розподіл потенціалу в
прошарку між двома паралельними
однойменно зарядженими поверхнями. В
достатньо розбавленому розчині
електроліту біля зарядженої поверхні
потенціал падає від значення
![]() (на межі адсорбційного шару) до нуля на
нескінченно великій відстані від неї
(пунктирні криві на рис.6.22). При зближенні
заряджених поверхонь до відстані, яка
відповідає товщині йонної атмосфери
(на межі адсорбційного шару) до нуля на
нескінченно великій відстані від неї
(пунктирні криві на рис.6.22). При зближенні
заряджених поверхонь до відстані, яка
відповідає товщині йонної атмосфери
![]() ,
відбувається зміна розподілу потенціалу
в прошарку між поверхнями, і в середині
прошарку з’являється мінімум потенціалу
(суцільна крива на рис.6.22). Потенціал в
середині прошарку
,
відбувається зміна розподілу потенціалу
в прошарку між поверхнями, і в середині
прошарку з’являється мінімум потенціалу
(суцільна крива на рис.6.22). Потенціал в
середині прошарку
![]() дорівнює
подвійному значенню потенціалу одиничного
дифузного шару на тій же відстані h/2 від
поверхні
дорівнює
подвійному значенню потенціалу одиничного
дифузного шару на тій же відстані h/2 від
поверхні![]() :
:
![]() .
(6.31)
.
(6.31)
|
|
|
Рис.6.22.
Зміна потенціалу
між поверхнями заряджених часточок |

В підсумку електролітична складова розклинювального тиску може бути наближено виражена наступним чином:
![]() ,
(6.32)
,
(6.32)
де с
– концентрація йонів у розчині; R –
газова стала; Т – абсолютна
температура; γ - стала, яка визначається
потенціалом поверхні
![]() ;
h – відстань між часточками;
;
h – відстань між часточками;
![]() - величина, обернена товщині дифузного
шару, що включає в себе концентрацію
електроліту; е – основа натурального
логарифму.
- величина, обернена товщині дифузного
шару, що включає в себе концентрацію
електроліту; е – основа натурального
логарифму.
Сили притягання – це сили Ван-дер-Ваальса , які складаються із сил дисперсійної, орієнтаційної та індукційної взаємодії. Роль дисперсійної взаємодії особливо суттєва в дисперсних системах, де кожна часточка представляє собою мікрооб’єм конденсованої фази, розміри якого великі порівняно з молекулярними. В цьому випадку вже на помітних відстанях відбувається часткова компенсація дисперсійної взаємодії, тобто часткове „насичення” поверхневих сил.
Для двох часточок з тонким плоским прошарком рідини товщиною h, енергія молекулярного притягання описується виразом:
![]() (6.33)
(6.33)
де А – константа Гамакера.
Від’ємний знак розклинювального тиску свідчить про намагання фаз до наближення під дією сил молекулярного притягання.
Згідно теорії ДЛФО загальний розклинювальний тиск ПЗАГ дорівнює сумі позитивної складової розклинювального тиску Пе (відштовхування) і негативної складової розклинювального тиску ПМ (молекулярного притягання):
ПЗАГ = Пе + ПМ . (6.34)
Оскільки енергія електростатичного відштовхування Uе та молекулярного притягання UМ є функцією від відстані h,то
![]() ,
(6.35)
,
(6.35)
![]() .
(6.36)
.
(6.36)
Після інтегрування рівнянь енергію взаємодії колоїдних часточок визначають за виразом:
![]() (6.37)
(6.37)
де В
– множник, який залежить від значення
електричних потенціалів ПЕШ, температури,
властивостей середовища, е – основа
натурального логарифму;
![]() - величина, зворотна товщині дифузного
шару; h – відстань між часточками;
А – стала Гамакера.
- величина, зворотна товщині дифузного
шару; h – відстань між часточками;
А – стала Гамакера.
На рис.6.23 показані потенціальні криві взаємодії колоїдних часточок дисперсних систем.
|
|
|
Рис.6.23. Потенціальні криві взаємодії колоїдних часточок: 1 – енергія відштовхування; 2 – енергія притягання; 3 – результуюча крива |

Міжмолекулярні сили притягання між часточками починають виявлятися на великих відстанях. Ця взаємодія за степеневим законом Дерягіна зростає зі зменшенням відстані h (рис.6.23, крива 2). Сили електростатичного відштовхування виникають тільки при перекриванні ПЕШ часточок, що наблизились одна до одної під дією сил притягання. Зі зменшенням відстані між часточками ці сили збільшуються за експоненціальною залежністю (рис.6.23, крива 1).
Результуюча потенціальна крива 3 одержана
з перших двох геометричним складанням
ординат. На малих відстанях при h![]() 0
сили електростатичного відштовхування
постійні, а сили міжмолекулярного
притягання максимальні. По мірі зростання
відстані h між часточками переважає дія
сил міжмолекулярного притягання, а сили
електростатичного відштовхування
переважають на середніх відстанях. На
результуючий кривій спостерігаються
два мінімуму. Перший мінімум (І) відповідає
безпосередньому контакту і злипанню
часточок, а другий мінімум (ІІ) – їх
взаємного притяганню у прошарку
середовища. На середніх відстанях на
кривій спостерігається максимум, який
характеризує потенціальний бар’єр, що
заважає зближенню часточок. Тобто на
цій відстані переважають сили
електростатичного відштовхування, які
перешкоджають злипанню часточок.
0
сили електростатичного відштовхування
постійні, а сили міжмолекулярного
притягання максимальні. По мірі зростання
відстані h між часточками переважає дія
сил міжмолекулярного притягання, а сили
електростатичного відштовхування
переважають на середніх відстанях. На
результуючий кривій спостерігаються
два мінімуму. Перший мінімум (І) відповідає
безпосередньому контакту і злипанню
часточок, а другий мінімум (ІІ) – їх
взаємного притяганню у прошарку
середовища. На середніх відстанях на
кривій спостерігається максимум, який
характеризує потенціальний бар’єр, що
заважає зближенню часточок. Тобто на
цій відстані переважають сили
електростатичного відштовхування, які
перешкоджають злипанню часточок.
Якщо середня кінетична енергія часточок більша ніж енергія, яка відповідає потенціальному бар’єру, то вони можуть подолати електростатичні сили відштовхування і наблизитися на дуже малу відстань (область мінімуму 1), де превалюють сили притягання, і злипнутися у агрегат. Агрегати, що утворилися, внаслідок розриву сольватних прошарків набувають деяких властивостей твердого тіла, тобто утворюється осад.
Якщо агрегатний бар’єр дуже високий і його колоїдні часточки не можуть подолати, то в такій системі агрегати не утворюються. Це випадок агрегативно стійкої системи.
Другий потенціальний мінімум (ІІ) відповідає взаємодії часточок на далеких відстанях одна від одної. Внаслідок дії сил відштовхування, які не дають часточкам наближатися дуже близько і коли їм заважають розійтися сили притягання, утворюються структуровані системи–гелі. Оскільки енергія взаємного притягання часточок в таких системах досить слабка, то ці структури легко руйнуються при струшуванні, тобто відбувається перехід гелю в золь. Через деякий час золь може знову переходити в гелеподібний стан.
Явище ізотермічного оборотного переходу
Золь L Гель,
що відбувається під впливом механічної дії (перемішування, струшування, вібрації, ультразвуку тощо) називається тиксотропією.
Коагуляція колоїдних систем електролітами відбувається за двома механізмами – концентраційними і нейтралізаційними (адсорбційними).
Концентраційна коагуляція пов’язна
зі збільшенням концентрації електроліту,
який є індиферентним і не спроможний
до специфічної адсорбції. Збільшення
концентрації індиферентного електроліту
в дисперсній системі призводить до
стискування дифузної частини ПЕШ.
Внаслідок цього частина протийонів
дифузного шару переходить в адсорбційний
шар, що стає причиною значного зменшення
![]() -потенціалу.
Одночасно введення електроліту збільшує
йонну силу розчину, пригнічує дифузію
протийонів і зменшує розпушеність
дифузного шару, що є впливовим фактором
для колоїдних систем із сильно зарядженими
часточками.
-потенціалу.
Одночасно введення електроліту збільшує
йонну силу розчину, пригнічує дифузію
протийонів і зменшує розпушеність
дифузного шару, що є впливовим фактором
для колоїдних систем із сильно зарядженими
часточками.
Нейтралізаційна коагуляція спостерігається
в дисперсних системах із слабо зарядженими
часточками, які мають низьке значення
поверхневого потенціалу. Коагуляцію
викликають йони, здатні до специфічної
адсорбції на поверхні часточок, і які
заряджені протилежно до них. Адсорбуючись,
ці йони знижують поверхневий потенціал,
паралельно знижується і
![]() -потенціал,
що призводить до наближення і злипання
колоїдних часточок. Поріг нейтралізаційної
коагуляції обернено пропорційний
квадрату величини заряду йона-коагулятора
(правило Ейлера-Кофе):
-потенціал,
що призводить до наближення і злипання
колоїдних часточок. Поріг нейтралізаційної
коагуляції обернено пропорційний
квадрату величини заряду йона-коагулятора
(правило Ейлера-Кофе):
![]() (6.38)
(6.38)
На рис.6.24 приведені потенціальні криві взаємодії колоїдних часточок при концентраційній і нейтралізаційній коагуляціях.
|
|
|
Рис.6.24. Потенціальні криві взаємодії колоїдних часточок при концентраційній (а) і нейтралізаційній коагуляціях: 1 – результуюча потенціальна криіва за відсутності коагулюючого електроліту; 1’ – при наявності електроліту; 2 – енергія відштовхування за відсутності коагулюючого електроліту; 2’ – при наявності коагулюючого електроліту |

Кінетика коагуляції. Кількісною
мірою процесу коагуляції є швидкість
коагуляції, яка характеризується зміною
чисельної концентрації часточок за
одиницю часу
![]() :
:
![]() (6.39)
(6.39)
На рис.6.25 показана залежність швидкості коагуляції від концентрації електроліту.
|
|
|
Рис.6.25. Залежність початкової швидкості коагуляції від концентрації електроліту: І – зона стійкого золю; ІІ – зона повільної коагуляції; ІІІ – зона швидкої коагуляції, СК.П і СК.Ш – поріг повільної і швидкої коагуляції |

Графічну залежність можна поділити на три частини:
І – зона стійкого золю, коагуляція не відбувається;
ІІ – зона повільної коагуляції, швидкість визначається як числом ефективних зіткнень, так і впливом різних факторів, які впливають на висоту потенціального бар’єра між часточками, що наближаються; зі збільшенням концентрації даного електроліту швидкість коагуляції збільшується;
ІІІ – зона швидкої коагуляції, коагуляція визначається тільки кількістю зіткнень і не залежить від інтенсивності факторів, що впливають на взаємодію між часточками, зокрема від концентрації електроліту.
Згідно з теорією швидкої коагуляції М.Смолуховського (1917 р.) процес коагуляції описується кінетичним рівнянням хімічної реакції другого порядку:
![]() (6.40)
(6.40)
Після інтегрування рівняння в межах
від
![]() до
до
![]() дістанемо:
дістанемо:
![]() (6.41)
(6.41)
де К
= 4![]() DR;
D – коефіцієнт дифузії; R – радіус
сфери дії сил притягання.
DR;
D – коефіцієнт дифузії; R – радіус
сфери дії сил притягання.
При умові половинної коагуляції (θ) рівняння набуває виразу:
![]() (6.42)
(6.42)
Величина θ для кожної дисперсної системи є постійною, тому її застосовують для характеристики дисперсних систем.
Коагуляцію дисперсних систем можна викликати і за допомогою суміші електролітів. При коагуляції сумішу електролітів спостерігаються три ефекти: адитивність, антагонізм і синергізм електролітів (рис.6.26).
Адитивність суміші електролітів полягає
в тому, що кожний електроліт діє окремо
(рис.6.26, пряма 1). Якщо пороги коагуляції
електролітів дорівнюють
![]() і
і
![]() ,
а їх концентрації С1 і С2,
то адитивність такої суміші характеризується
рівнянням:
,
а їх концентрації С1 і С2,
то адитивність такої суміші характеризується
рівнянням:
![]() (6.43)
(6.43)
На графіку (рис.6.26) пряма 1 сполучає
значення порогів коагуляції
![]() і
і
![]() кожного окремого електроліту. Адитивність
спостерігається у йонів однакового
заряду і близьких за властивостями ( K+
і Na+, Cl- і Br
-).
кожного окремого електроліту. Адитивність
спостерігається у йонів однакового
заряду і близьких за властивостями ( K+
і Na+, Cl- і Br
-).
|
|
|
Рис.6.26. Схема дії двох електролітів при коагуляції: 1 – адитивність; 2 – антагонізм; 3 – синергізм;
|

При антагонізмі електролітів вони протидіють один одному, і для досягнення коагуляції дисперсної системи необхідно більше відповідної частки кожного з них, ніж з правилом адитивності (рис.6.26, крива 2). Приклади антагонізму є коагуляція AgJ сумішу Al(NO3)3 і K2SO4.
Синергізмом називають взаємне посилення коагулюючої дії одного електроліту при додаванні другого. Для досягнення коагуляції дисперсної системи їх потрібно менше, ніж це необхідно за правилом адитивності (рис.6.26, крива 3). Синергізм виявляється, наприклад, при дії суміші LiCl і CaCl2 на колоїдний розчин HgS.
Коагуляція може відбуватися і при взаємодії двох стійких колоїдних систем, в яких часточки мають протилежні заряди. Такі колоїдні системи виявляють максимальний вплив один до одного, якщо сумарний заряд їх часточок дорівнює нулю. Причиною взаємної коагуляції (гетерокоагуляції) є електростатичне притягання.
Окремий випадок цього явища спостерігається при взаємодії однойменно заряджених колоїдних систем, яку пояснюють хімічною і адсорбційною взаємодією.
Часточки дисперсних систем взаємодіють також з твердим тілом, поверхня якого має протилежний за знаком заряд. Утворення коагулянту на таких тілах називається гетероадагуляцією.
Наряду з розглянутими вище механізмами коагуляції важливе значення має і так звана ортокінетична коагуляція. Так при седиментації крупні часточки, що рухаються з великою швидкістю, можуть доганяти повільно осідаючі часточки і „захоплювати” їх. Осідаюча крупна часточка здатна захопити більш дрібні, центр яких знаходиться в деякому прямовисному циліндрі з віссю, що проходить через центр більш крупної часточки; основа цього циліндру називається перерізом захоплювання (рис.6.27). Самі дрібні часточки дифундують по поверхні більш крупної часточки внаслідок обтікання її рідиною; наближення часточок обумовлено дією інерційних сил, які виникають на початку обтікання (J).
|
|
|
Рис.6.27. Схема ортокінетичної коагуляції: r1 - радіус крупної осідаючої часточки; r2 – радіус мілкої седиментуючої часточки |
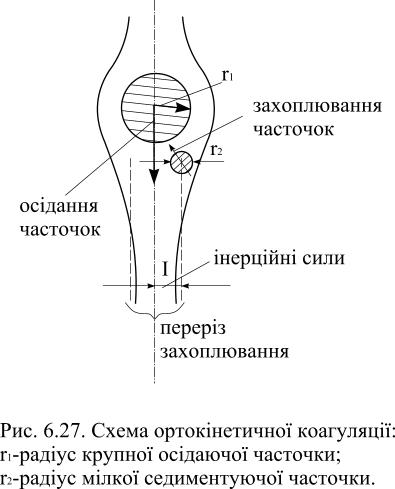
Явище взаємної коагуляції широко використовується для руйнування колоїдних систем, зокрема в нафтодобувної промисловості, для очищення ґрунтових вод, промислових стоків.
Гетрокоагуляція і ортокінетична коагуляція лежать в основі ґрунтоутворюючих процесів; з їх протіканням пов’язані процеси водоочистки, пінної флотації, вловлювання пилу, аерозолів, штучного утворення осадів з хмар, тощо.
Порушення стійкості дисперсних систем під впливом розчинів ВМС. Дисперсні системи, до яких додаються розчини ВМС, в значній мірі втрачають стійкість, тобто сенсибілізують. Поріг коагуляції таких систем менший, ніж для вихідних. Це пов'язано з адсорбцією макромолекули полімеру одночасно на поверхні декількох часточок дисперсної системи. Утворюючи між часточками дисперсної фази „містки” макромолекули полімеру настільки порушують стійкість дисперсної системи, що вона утворює об’ємний пухкий осад. Така адсорбційна коагуляція називається флокуляцією. Частинки осаду-флокули добре фільтруються тому, що при взаємодії адсорбційних шарів полімерів між ними виникають сили відштовхування. Внаслідок цього проміжні ділянки макроланцюга полімеру між адсорбованими ділянками перебувають у розчині подалі від поверхні часточок.
Сили відштовхування мають ентропійну природу. Таке ентропійне відштовхування називають стеричним захистом. Він діє коли відстань між поверхнями часточок не перевищує подвійну товщину адсорбційного шару полімеру.
Стійкість розчинів ВМС. Порушення стійкості розчинів ВМС досягається шляхом погіршення їх розчинності. Для цього змінюють температуру, додають розчинники або електроліти. При цьому відбувається виділення ВМС з розчину. Цей процес називають висолюванням.
Дослідженнями встановлено, що на висолювання гідрофільних полімерів впливають як аніони, так і катіони. Висолююча дія аніонів відображається рядом (ряд Гофмейстера):
![]()
Катіони мають менший висолюючий ефект, їх можна розмістити в такий ліотропний ряд:
![]() .
.
Висолююча дія йонів залежить від їх зарядів, якщо концентрації цих йонів однакові. Це пояснюється більшою гідратацією йонів з більшою величиною заряду порівняно з меншою.
На висолювання високомолекулярних сполук значно впливають рідини, які добре розчиняються в дисперсійному середовищі, і практично не розчиняють полімер. Приклади такої рідини є етиловий спирт. За допомогою спирту і суміші спирту з етером висолюються практично повністю більшість гідрофільних полімерів.
Явище висолювання характеризується оборотністю. Оборотність цього явища полягає в тому, що після видалення з осаду полімеру електроліту полімер знову може розчинятися з поновленням основних властивостей, які мав розчин до висолювання.
Під час змішування розчинів ВМС з розчинами електролітів, з ацетоном, спиртом, з розчинами інших ВМС тощо, можуть утворюватись краплі рідкого ВМС. З часом краплі зливаються і у вигляді концентрованого розчину ВМС осідають на дно, а зверху залишається шар більш розведеного розчину ВМС. Концентрований шар ВМС називають коацерватом, а явище, при якому утворюється коацерват, називається коацервацією.
Коацервацію вважають незакінченим, початковим процесом висолювання, при якому міцели втрачають дифузний шар і зберігають гідратний шар, молекули якого з’єднуються в рідкі краплі коацервату. Наприклад, якщо до розчину альбуміну поступово добавляти концентрований розчин Na2SO4, утворюється коацерват альбуміну.
Явище коацервації широко використовується для фракціонування природних і синтетичних високомолекулярних сполук.
Грубодисперсні системи. Природа стійкості багатьох систем в значній мірі залежить від фазового стану дисперсної системи. Дисперсні системи з різним агрегатним станом: аерозолі, суспензії, емульсії і піни відносяться до грубодисперсних систем. Часточки цих систем більші за розміром ніж колоїдні.
Деякі біологічні системи – мікроорганізми, синьо-зелені водорості, спори рослин, тваринні і рослинні мембрани тощо, за своїми властивостями також відносяться до грубодисперсних систем.
Піни. Піна – дисперсія газу або повітря в рідкому або твердому середовищі. Тому існують як рідкі так і тверді піни. Піни представляють собою типову ліофобну систему. Розрізняють розбавленні дисперсії газу в рідині, які називаються газовими емульсіями. Такі піни містять більше 70% газової фази в об’ємі піни. У концентрованих пінах газоподібна фаза є основною (до 99% об’єму) частиною системи. В якості характеристики концентрації піни користуються відношенням об’єму піни до об’єму рідини, що міститься в ній. Цю величину називають кратністю піни. За цією характеристикою піни поділяються на розбавлені і концентровані.
|
|
|
Рис.6.28. Схеми будови комірок пін: а – сферичної; б – п’ятикутної |

В пінах комірки, які заповнені газовою фазою (повітрям) розділені тонкими плівками дисперсійного середовища (рис.6.28). Комірки можуть мати правильну сферичну форму (рис.6.28, а), а при стикуванні комірок вони набувають форму багатогранників (рис.6.28, б). Характерною коміркою пін є пентагональний додекаєдр. Це дванадцатигранник з п’ятикутними гранями, що має 30 ребер і 20 вершин (рис.6.29). Ребрами пінної комірки є канали Плато, які заповнені дисперсійним середовищем. В одному каналі такої комірки можуть збігатися тільки три плівки, які розташовані під кутом 1200.
|
|
|
Рис.6.29. Комірка пентагонального додекаедру |

Площа перерізу каналу Плато має форму „трикутника з увігнутими боками”.
Вершини сусідніх пентагональних додекаедрів утворюють вузли, в яких збігаються чотири канали. Канали і вузли утворюють єдину розгалужену систему в якій відбувається перенесення дисперсійного середовища, а також її стікання під дією сил тяжіння.
В реальних системах на початку утворення піна може бути монодисперсною, але внаслідок розриву плівок стає полідисперсною. При цьому завжди зберігається правило Плато: три плівки утворюють канал, чотири канали утворюють вузол (вершину).
Утворення стійких пін можливе тільки при наявності стабілізаторів-піноутворювачів. Піноутворювачами є ПАР, які поділяють на два типи: 1) першого роду – зменшують величину поверхневого натягу (нижчі спирти і кислоти); 2) другого роду – на поверхні розподілу утворюють міцні адсорбційні драглеподібні плівки (мила, алкалоїди, таніни, деякі барвники, білки, карбометилцелюлоза тощо).
Піни, що утворюються за допомогою піноутворювачів, мають велику механічну міцність структурованих адсорбційних шарів. Стінки комірок таких пін можна розглядати як такі, що утворені двома двомірними конденсованими (твердими) плівками.
Рідини, які здатні тверднути, утворюють тверді піни (пемза, пінопласт, пінополістирол тощо).
Піни широко використовуються в різних областях сучасної техніки: при гасінні пожеж (особливо нафтових родовищ, продуктів її крекінгу), при флотаційному збагаченні руд, у виробництві харчових і кондитерських продуктів (хліб – це приклад твердої піни), теплоізоляційних матеріалів (мікропориста гума, пінобетон, пориста цегла).
Добування пін, як правило, відбувається шляхом диспергування повітря або газів в рідині, яке містить ПАР. Диспергування газу відбувається в шарі рідини (барботажні піни) або за допомогою мішалок в об’ємі рідини. Також застосовують піногенератори різних конструкцій. Утворення піни в них відбувається на сітках, що встановлені на шляху газорідинного потоку. Такі піногенератори забезпечують утворення великої кількості піни необхідної кратності, що дозволяє швидко гасити пожежі і особливо горіння різних речовин.
Піни одержують також конденсаційним методом, при якому відбувається виділення будь-якого газу, що утворюється за рахунок хімічної реакції або біохімічного процесу. Так у вогнегасниках відбувається хімічна реакція:
![]()
при виробництві газобетону:
![]()
а випіканні хліба:
![]()
Руйнування пін супроводжується зміною параметрів, що характеризують будову і властивості пін. Тобто відбувається потоншення і розрив плівок комірок, ізотермічна перегонка газової фази від дрібних комірок до більш крупних, а також синерезису – витікання дисперсійного середовища (рідини) із каналів Плато під дією сил тяжіння.
В багатьох випадках піноутворення є небажаним явищем. Тому руйнування (гасіння) пін або припинення піноутворення є важливою задачею у багатьох технологічних процесах. Так, велика кількість рясної піни може утворюватися в хімічних процесах, у мікробіологічному виробництві, що пов’язано з виділенням газів або їх взаємодією з рідинами, надмірна кількість піни утворюється при кипінні рідин в котлах, при роботі пральних машин, мішалок. У цих випадках гасіння пін досягається використовуванням ПАР, які більш поверхнево-активні ніж ті, що наявні в початковій системі піноутворення. Такі ПАР сприяють самовільному руйнуванню пін.
Піни, що утворюються, можна руйнувати за допомогою різних методів: дією перегрітої пари, ультразвуку, або введенням піногасників. Піногасники – це ПАР, які мало розчиняються у воді, але швидко розтікаються на її поверхні у вигляді мономолекулярного шару. Вони витискають піноутворювачі з поверхневого шару плівки і тим самим на декілька порядків знижують стійкість пін. Піногасниками можуть бути середні гомологи спиртів , поліаміди жирних кислот, рідкі жирні кислоти і їх гліцериди, пропіленгліколі і їх похідні, силіконові масла.
Емульсії. Емульсіями називаються дисперсії рідин в рідких дисперсійних середовищах, які взаємно нерозчинні. У зв’язку з цим часточки дисперсної фази і дисперсійного середовища повинні різко відрізнятися за полярністю. Тому особливістю емульсій є можливість утворення емульсій двох типів: прямої і оберненої. Більш полярною рідиною звичайно є вода (В), малополярну рідину прийнято називати „масло” (М). В зв’язку з цим пряму емульсію позначають М/В (емульсії І роду), а обернену – В/М (емульсії ІІ роду). Можливі також так звані „множинні емульсії”, в яких дисперсійне середовище частково дисперговане в краплинах дисперсної фази.
Емульсії за концентрацією дисперсної фази поділяють на три групи: 1) розведені (з концентрацією дисперсної фази не вище 0,1% об’єму емульсії); 2) концентровані (з концентрацією дисперсної фази 0,1-70% об’єму); 3) висококонцентровані (з вмістом дисперсної фази вище 70% об’єму емульсії). Висококонцентровані емульсії за структурою і властивостями подібні пінам, тому їх іноді називають спумоїдними (піноподібними) емульсіями (рис.6.30).
|
|
|
Рис.6.30. Схематичне зображення емульсії: а – розбавленої; б – висококонцентрованої |

Стійкість емульсій, у першу чергу, залежить від концентрації дисперсної фази. В більш стійкої розведеної емульсії краплі рідини (які є монодисперсними (рис.6.30, а) зливаються одна з одною (коалесціюють), і система швидко розшаровується. Висококонцентровані емульсії менш стійкі. На відміну від розведених висококонцентровані емульсії мають частинки більших розмірів і різної форми (рис.6.30, б). Такі емульсії називають полідисперсними.
Утворення стійких емульсій можливе при застосуванні стабілізаторів, які називають емульгаторами. Це звичайні та високомолекулярні ПАР (окси- і сульфоспирти, солі і амінопохідні жирних кислот, амонієві основи, білки, сапонін, крохмаль, казеїн, желатина, полакриламід, поліоксиетилен та ін.). Емульгатор адсорбується на межі поділу фаз і при цьому зменшує питому вільну поверхневу енергію. Такий міжфазовий шар (МФШ) має будову, яка зображена на рис.6.31, б.
Якщо розчинна у воді і нерозчинна в маслі ПАР приводить до зниження поверхневого натягу на межі МФШ – вода утворюється емульсія типу М/В і навпаки, якщо ПАР розчинна в маслі утворюється емульсія типу В/М (рис. 6.31, а і в).
При певних умовах спостерігається обернення фаз емульсій, тобто коли емульсія даного типу при введенні будь-яких реагентів або при зміні умов перетворюється в емульсію протилежного типу.
|
|
|
Рис.6.31. Схеми форми міжфазового шару емульсій залежно від поверхневого натягу з боку „масляної” і водної фаз |

Щоб визначити тип емульсії застосовують різні методи. Так, додавання до емульсії фарби, яка розчиняється тільки в одній з рідин (водорозчинної або маслорозчинної) відповідне забарвлення має можливість визначити тип емульсій. Інколи застосовують метод розбавлення. Якщо емульсія легко розбавляється водою, то вода є дисперсійним середовищем, тобто це емульсія типу М/В і навпаки емульсія типу В/М легко розбавляється „маслом”.
Тип емульсії можна встановити також кондукторометричним методом. Якщо дисперсійним середовищем є вода, електропровідність якої висока, то це емульсія типу М/В. В той же час електрична провідність емульсій типу В/М дуже низька.
Під дією ультрафіолетового випромінювання в темряві емульсії В/М флуоресціюють на відміну емульсій типу М/В, які забарвлення звичайно не набувають.
Легко встановити тип емульсії нанесенням краплини емульсії на фільтруваний папір. Якщо пляма швидко розпливається, а в центрі залишається невелика краплинка („масло”), то це емульсія типу М/В.
Тип емульсії залежить від властивостей поверхні емульгатора. За правилом Банкрофта рідина в якої розчиняється емульгатор вважається дисперсійним середовищем.
Це означає, що гідрофільні емульгатори сприяють утворенню емульсій типу М/В, а гідрофобні – типу В/М. Механізм стабілізуючої дії емульгаторів полягає в утворенні на частинках дисперсної фази захисних оболонок, які запобігають їх зливанню. Їх дія залежить від співвідношення гідрофільних і ліпофільних ділянок молекули емульгатора. Це співвідношення називають гідрофільно-ліпофільним балансом (ГЛБ).
Значення ГЛБ для деяких молекул ПАР приведені в табл.6.5.
Таблиця 6.5
Значення деяких ПАР
|
ПАР |
ГЛБ |
|
Олеїнова кислота |
1,0 |
|
Бутанол |
7,0 |
|
Олеат натрію |
18,0 |
|
Лаурилсульфат натрію |
40,0 |
Великі значення ГЛБ відповідають гідрофільним ПАР, що стабілізують прямі емульсії. Навпаки, малі значення ГЛБ характерні для олеофільних ПАР, що стабілізують обернені емульсії.
Найбільшу емульгуючу дію мають мила лужно-земельних металів, які стабілізують емульсії типу В/М і лужні мила, що стабілізують емульсії типу М/В (рис.6.32).
|
|
|
Рис.6.32. Схема стабілізації емульсії В/М (а) і емульсіх М/В лужним милом (б) |

Наочним прикладом утворення структурно-механічного бар’єру, що є сильним фактором стабілізації дисперсій, може розглядатися стабілізація емульсій тонко подрібненими порошками. У випадку крапель води, що покриті гідрофобним порошком, наприклад вугіллям, в масляній фазі вода відтискується з прошарків між часточками внаслідок гідрофобності вугілля, і при зіткненні краплини води не можуть увійти в безпосередній контакт (рис.6.33, а). Навпаки, гідрофільний порошок, наприклад крейда (СаСО3) захищає собою масляну фазу і не дозволяє увійти в контакт краплинам масла в водному дисперсійному середовищі (рис.6.33, б).
У більшості випадків емульсії одержують диспергуванням у спеціальних апаратах – гомогенізаторах, або в колоїдних млинах.
Застосовують також ультразвукове диспергування, конденсацію частинок дисперсної фази у воді, протискання рідини крізь сита з дуже малими отворами під тиском. Найпростіший спосіб – це інтенсивне перемішування або збовтування рідин. У всіх випадках необхідна присутність відповідного емульгатора.
|
|
|
Рис.6.33. Схема стабілізації емульсій гідрофобними (а) і гідрофільними (б) порошками |

Найважливішим представником природних емульсій є сира нафта, яка містить – 6-% засоленої води і дуже сильно стабілізована природними ПАР і смолами. Широке застосування мають емульсії в техніці й хімічній технології: це процеси механічної обробки з застосуванням емульсійних мастило-охолоджувальних рідин, емульсійної полімеризації, переробки і добування харчових продуктів (молоко, вершкове масло, майонез, маргарин) і фармацевтичних препаратів для внутрішнього вживання і як зовнішні засоби.
Процеси руйнування емульсій наближені за природою і механізмами протікання до процесів руйнування пін. В розбавлених емульсіях відбувається седиментація краплин: вгору – при меншій густині дисперсійної фази (наприклад, утворення вершків) або вниз – в зворотньому випадку.
На процес седиментації може накладатися агрегування краплин емульсії. Це явище називається флокуляцією. Флокуляція приводить до збільшення ефективного розміру агрегатів, що осідають, а також до збільшення швидкості їх осідання.
Велика увага приділяється розробці методів руйнування емульсій (деемульгування). Так, руйнування емульсії сирої нафти є першою і достатньо важкою стадією переробки нафти. Часто емульсії руйнують центрифугуванням, додаванням електролітів, сильних реагентів (кислот), ПАР, нагріванням, заморожуванням або накладанням електричного поля.
Драглі. Дисперсні системи, а також розчини ВМС, які здатні желатинуватись, тобто створювати драглисту масу з властивостями напіврідинного, напівтвердого тіла, називаються драглями або гелями.
Коагуляція седиментаційно-стійких вільнодисперсних систем (золів) може не супроводжуватися наочним седиментаційним розшаровуванням системи. Це постерігається в тих випадках, коли агрегування часточок призводить до розчинення суцільної просторової сітки часточок, які заповнюють весь об’єм системи, тобто до утворення зв’язанодиперсної системи – гелю. В зв’язанодисперсних системах часточки можуть залишатися розділеними прошарками дисперсійного середовища, або може відбутися повне витіснення останнього. При цьому відбувається безпосередній контакт для твердих часточок і повне зливання – для краплин і пухирців.
Прикладом некристалічних конденсаційних дисперсних структур є силікати (сикагелі). Силікагелі утворюються при виділенні нової аморфної фази при взаємодії силікату натрію з кислотою:
Na2SiO3
+ 2HCl + H2O
![]() 2NaCl + Si(OH)4,,
2NaCl + Si(OH)4,,
(HO)3
![]() Si
– OH + HO – Si
Si
– OH + HO – Si
![]() (OH)3
+ …
(OH)3
+ …
![]()
(OH)3![]() Si
– [- O – Si(OH)2 -]n
– O – Si
Si
– [- O – Si(OH)2 -]n
– O – Si
![]() (OH)3 + (n-1)H2O.
(OH)3 + (n-1)H2O.
За спільними характеринми властивостями гелі поділяють на: ліогелі, які містять велику кількість рідини (кисіль, кисле молоко, холодець, драглі силікатної кислоти тощо); ксерогелі – висушені ВМС (желатина, целюлоза, шкіра, шерсть, каучук, ріг тощо); аерогелі – високопоруваті ксерогелі (силікагель, вугілля).
Під впливом дії тих або інших факторів (старіння, температура, механічної дії) ряд гелів змінює гідрофільність та інші властивості. Так, борошно має здатність зв’язувати воду, утворюючі тісто, яке при випіканні досягає максимальної гідрофільності (з 44 % до 82,7 %). Під час зберігання хліба зв'язана вода поступово втрачається, тобто відбувається старіння (черствіння) хліба, який містить 67,4 % води.
При утворенні драглів відбувається процес гелеутворення, тобто желатинування. Желатинування є процесом переходу колоїдного розчину (або полімеру) в драглі, який відбувається без відокремлення дисперсної фази від дисперсійного середовища. Разом з цим не відбувається порушення того співвідношення їх, яке було у вихідному золі. Желатинування обмежується об’ємним захопленням дисперсійного середовища часточками дисперсної фази (іммобілізацією). Процес коагуляції і желатинування пов’язаний з структуроутворенням подовженої форми часточок дисперсної фази, яке супроводжується їх певною орієнтацією і взаємодією. Ці часточки з’єднуються мж молекулами або хімічними силами зв’язку і створюють механічну стійку структуровану сітку, яка рівномірно заповнює зайнятий розчином простір (рис.6.34). Утворення гелю проходить через ряд послідовних стадій:
|
|
|
Рис.6.34 |



Si(OH)4![]() Колоїдні
часточки (золь)
Колоїдні
часточки (золь)
![]() Сітка
часточок (драглі)
Сітка
часточок (драглі)
![]()
Розбавлений
золь
![]() Слабкі
драглі або осад
Слабкі
драглі або осад
![]()
Концентрований
золь
![]() Міцні
тверді драглі
Міцні
тверді драглі
![]()
Вологі
драглі
![]() Висушені драглі,
Висушені драглі,
або
Структуроутворювання
![]() Желатинування
Желатинування
![]()
![]() Дозрівання
Дозрівання
![]()
![]() Синерезис
Синерезис
Драглі з розчинів полімерів утворюються при додаванні електролітів або при їх випарюванні, охолодженні, обмеженому набуханні полімеру у низькомолекулярній рідині:
Додавання електроліту;
Концентрування;
Охолодження;
В
 МС
МС
![]() Розчин ВМС
Розчин ВМС
![]() Драглі
Драглі
О бмежене
набухання
бмежене
набухання
За певних умов (струшування, перемішування, вібрація, нагрівання) внутрішня структура часточок драглів руйнується і вони переходять в розчин (вільнодисперсну систему). Після припинення механічної дії під впливом теплового руху часточок і сил міжмолекулярної взаємодії ця структура знову відновлюється. Явище механічного руйнування драглів і наступного переходу золю в гель, називається тиксотропією. Цей процес можна зобразити схемою:
|
Механічна дія
Г Нагрівання
|

 ель
Золь
ель
ЗольПроцес утворення драглів дуже поширений у природі. Багато складних біологічних і фізико-хімічних процесів супроводжуються желатинуванням. Через стадію желатинування утворюються гелі грунтів і осадові породи (каолін, глинозем тощо), в живих організмах і рослинах утворюються крохмаль, целюлоза, білкові речовини. Перебувають у стадії желатинування різноманітні харчові продукти (желе, мармелад, джем, сир, кисле молоко, плодові та овочеві соки, кисіль, холодець, хліб тощо).
Аерозолі. Дисперсні системи з газуватим дисперсійним середовищем, незалежно від агрегатного стану дисперсної фази (рідкої або твердої), називаються аерозолями. Системи з рідкою дисперсною фазою – це тумани, з твердою – дими, пил і порошки, якщо одночасно вони містять і рідкі і тверді часточки – смоги. В смогах рідка фаза конденсується на поверхні твердих часточок. Такі аерозолі частіше всього присутні в атмосфері великих промислових міст.
Тумани іноді називають фогом (імла), якщо вони достатньо густі і сильно погіршують видимість. Летку золу, продукти неповного згоряння або їх суміш (як тверді, так і рідкі) називають кіптявою або смоком.
Смог – це штучне слово, яке складене зі слів смок і фог, що використовується для визначення любого небажаного забруднення атмосфери. Існують два типи смогів – лос-анжелеський і лондонський; лос-анжелеський смог має фотохімічну природу і обумовлений випускними газами автомобілів, лондонський – обумовлений неповним згорянням вугілля і характеризується відносно високим вмістом диоксиду сульфуру і твердих часточок.
|
|
|
Рис.6.35. Класифікація атмосферних забруднень за розмірами та основні методи визначення розмірів частинок |

Прийнято рахувати аерозольними системами, що містять часточки розміром 10-9 – 10-3 м. Системи з більш крупними часточками називають аерозавісами.
За розміром часточок, що складають
аерозольну систему, усі аерозолі
поділяють на високодисперсні (діаметр
часточок менш ніж 10 нм), тонко дисперсні
(10![]() d
d![]() 100
нм) і грубо дисперсні (діаметр часточок
більш ніж 100 нм). На рис.6.35 приведена
класифікація атмосферних забруднювачів
за розміром їх часточок. В природі
аерозолі обумовлюють практично усі
метерологічні, в тому числі грозові,
явища. В сільському господарстві з
використанням аерозолів пов’язані
проблеми штучного дощування, застосування
отрутохімікатів для боротьби зі
шкідниками і хворобами рослин. В техніці
– це очищення повітря, різних технологічних
газових сумішей, вловлювання цінних
матеріалів з газів, що викідаються. В
биту і медицині – це застосування
аерозольної форми препаратів, ліків,
лаків, фарб, тощо. З проблемою захисту
від аерозолів, які утворюються під час
роботи атомних електростанцій, переробці
руд, при згорянні палива, при аваріях в
промисловості, вибухах військових
речовин, викидів вулканічних газів і
т.п., пов’язані ключові питання охорони
здоров’я людей і захисту навколишнього
середовища.
100
нм) і грубо дисперсні (діаметр часточок
більш ніж 100 нм). На рис.6.35 приведена
класифікація атмосферних забруднювачів
за розміром їх часточок. В природі
аерозолі обумовлюють практично усі
метерологічні, в тому числі грозові,
явища. В сільському господарстві з
використанням аерозолів пов’язані
проблеми штучного дощування, застосування
отрутохімікатів для боротьби зі
шкідниками і хворобами рослин. В техніці
– це очищення повітря, різних технологічних
газових сумішей, вловлювання цінних
матеріалів з газів, що викідаються. В
биту і медицині – це застосування
аерозольної форми препаратів, ліків,
лаків, фарб, тощо. З проблемою захисту
від аерозолів, які утворюються під час
роботи атомних електростанцій, переробці
руд, при згорянні палива, при аваріях в
промисловості, вибухах військових
речовин, викидів вулканічних газів і
т.п., пов’язані ключові питання охорони
здоров’я людей і захисту навколишнього
середовища.
Більшість специфічних властивостей
аерозолів пов’язані з особливостями
дисперсійного середовища – повітря,
його низькою в’язкістю і електропровідністю
(![]() =
8,85 . 1012 ф/м). Наявність зарядів
на поверхні аерозольних часточок
обумовлює ряд важливих явищ.
=
8,85 . 1012 ф/м). Наявність зарядів
на поверхні аерозольних часточок
обумовлює ряд важливих явищ.
Осідання аерозольних часточок (наприклад, краплин туману або дощу) обумовлює появу електричного поля з напругою Е порядку кВ на см. Ці значення Е можуть бути і більше, в цьому випадку можливий пробій повітря – блискавка.
Реальні аерозолі полідисперсні. Тому при їх седиментації спостерігається ортокінетична коагуляція часточок (захоплення крупними часточками більш мілких) (рис.6.36, а). Седиментуюча велика часточка, що утворилася, внаслідок опору повітря і сил гравітації витягується до тих пір поки не перевищується межа стійкості рідкого циліндра і ділиться на дві менших часточки (рис.6.36, б). Ці часточки, що утворилися знову збільшуються в процесі седиментації, а потім подрібнюються. Такий процес, що повторюється, прискорює швидкість седиментації аерозольних часточок до їх повного осідання.
|
а) |
б) |
|
Рис.6.36. Руйнування аерозолів: а) ортокінетична коагуляція; б) розкладання великої часточки на дві менших розмірів |
|

Такий механізм відбувається в атмосфері при руйнуванні туману, дощових хмар. Залежно від концентрації аерозольних часточок і температурних умов відбувається випадання води у вигляді роси, дощу, снігу чи граду. На практиці за таким механізмом руйнують грозові хмари, що несуть загрозу випадання граду і пошкодження сільськогосподарських угідь. Для цього у верхівку хмар за допомогою зенітних гармат доставляють зародкоутворювачі (кристали йодиду аргентуму, хлориду кальцію, твердий диоксиген карбону і деякі інші речовини колоїдного розміру), які викликають швидку конденсацію водяної пари і зростання крапель води (або кристаликів льоду в переохолоджених хмарах), що приводить до їх випадання. Аналогічним чином можна розсіювати тумани, використовуючи для цього авіатехніку.
Існуючі в атмосферному повітрі мікроорганізми, бактерії, віруси, пилок, насіння і спори рослин представляють собою так звані біологічні аерозолі, які переміщуються на величезні відстані і часто є носіямиінфекційних захворювань і епідемій.
Для аерозолів характерні такі специфічні явища, як термофорез та термопреципітація. Термофорез – переміщення аерозольних часточок у напрямку зниження температури, яке обумовлене тиском більш швидких молекул газу з „гарячого боку”. У великих часточках коли поверхня нагріта нерівномірно, то вздовж неї у напрямку менших температур виникає потік газу. Таке явище називають тепловий плин газу. Він породжує силу, яка діє вздовж поверхні часточки у тому ж напрямку, що зумовлює її рух у „холодний бік”.
Термопреципітація – осадження часточок аерозолю на холодних поверхнях. Взаємодія часточок з такою поверхнею приводить до зменшення кінетичної енергії часточок аерозолю.
При односторонньому освітленні часточок аерозолю вони можуть рухатися як у напрямку світлового променя, так і назустріч йому. Таке явище називається фотофорезом і є окремим випадком термофорезу. Так, у нижній стратосфері, де тиск повітря високий частинки рухаються вгору до Сонця. У верхній стратосфері тиск падає і часточки починають рухатися у протилежному напрямку – вниз. Отже, часточки рухаються в обмеженому шарі стратосфери. Це приводить до накопичення і утворення стійких шарів аерозолів, які впливають на прозорість стратосфери, а в цілому і на клімат.
В промисловості для руйнування аерозолів з метою очистки газових сумішей широко використовують дію електричного поля, фільтрування через тонковолокнисті фільтри, осадження в циклонах, електрофільтрах тощо.
Колоїдні ПАР. Карбонові ланцюги молекул ПАР в полярному розчиннику – воді об’єднуються в компактне ядро, а гідратовані полярні групи, що повернені в бік водяної фази, утворюють гідрофільну оболонку. Такий асоціат молекул ПАР (з числом молекул 20-100 і більше) є сферичною колоїдною часточкою (міцелою) (рис.6.37).
Міцелярні дисперсні ПАР виявляють властивості, які належать колоїдно-дисперсним системам: світлорозсіювання, електропро-відність, швидкість дифузії, в’язкість та ін.
Здатність до міцелоутворення володіють не усі ПАР, а тільки ті, які мають оптимальні співвідношення між гідрофобною і гідрофільною частинами, що визначається величиною ГЛБ. До міцелоутворюючих ПАР відносяться натрієві, калієві і амонієві солі жирних кислот з довжиною ланцюга С12-20, алкілсульфати, алкилбензолсульфонати та інші синтетичні йоногенні і нейоногенні ПАР.
|
|
|
Рис.6.37. Схема будови сферичних (а) і пластинчастих (б) міцел колоїдних пар |

При збільшенні вмісту ПАР в розчині вище деякої критичної концентрації спостерігається помітний зріст світлорозсіювання; ізотерми поверхневого натягу замість звичайного поступового зниження поверхневого натягу мають злом, а при подальшому зростанні концентрації значення поверхневого натягу залишається незмінним (рис.6.38). Аналогічно злом з’являється і на кривих концентраційної залежності питомої і еквівалентної електропровідності розчинів йоногенних ПАР. Концентрація, вище якої починається міцелоутворення, називається критичною концентрацією міцелоутворення (ККМ).
|
|
|
Рис.6.38. Залежність властивостей розчинів колоїдних ПАР від концентрації |

Дослідження водних дисперсій, що утворюють міцели ПАР, показали, що міцелоутворення як шляхом конденсації молекул в міцели, так і шляхом диспергування макрофаги може відбуватися тільки вище деякої температури ТК, яку називають точкою Крафта (рис.6.39 ). Нижче температури ТК розчинність ПАР мала і є менше ніж ККМ. В цієї області температур існує рівновага між кристалами мила і істинним розчином ПАР, концентрація якого зростає по мірі зростання температури. Тому в розчинах ПАР, для яких точка Крафта лежить в області вище 50 - 800 С, в звичайних умовах міцелоутворення не спостерігається.
|
|
|
Рис. 6.39. Залежність розчинності ПАР від температури |

Для усіх міцелоутворюючих ПАР ККМ лежить в області малих значень концентрацій (10-5 – 10-2 кмоль/м3. Зі зростанням вмісту ПАР в розчині (в десятки і сотні разів, що перевищує ККМ) сферичні міцели перетворюються в анізометричні еліпсоїдальні і циліндричні, а потім в паличкоподібні, стрічкові і пластинчасті міцели (рис.6.37). При цьому кожному значенню концентрації відповідає термодинамічна рівновага
Сферичні L Анізометричні L Стрічкові
міцели міцели міцели
Подібні системи, проміжкові між істинними рідинами і твердими тілами, часто називають рідкими кристалами (рис.6.37). Таким чином система ПАР – вода може при зміні вмісту компонентів переходить в різні стани, від гомогенної системи, через стадію ліофільної колоїдної системи, до макрогетерогенної системи. При цьому різним станам системи відповідає певна термодинамічна рівновага:
І
 стинний
L Сферичні L
Анізометричні L
Кристали L
Істиний
стинний
L Сферичні L
Анізометричні L
Кристали L
Істиний
розчин міцели в міцели розчин
розчині

 Колоїдна система
Колоїдна система
В неводних розчинах ПАР тобто в розчинах ПАР в карбонгідрогенних середовищах можуть утворюватися міцели з протилежною орієнтацією молекул (рис.6.40). При формуванні таких зворотних міцел полярні групи об’єднуються в гідрофільне (олеофобне) ядро, а карбонгідрогенні радикали, що повернуті в бік рідинного неполярного середовища, утворюють олеофільну оболонку, яка екранує внутрішню гідрофільну частину міцели від контакту з карбонгідрогенним середовищем.
|
|
|
6.40.Будова зворотної міцели |

Введення в систему ПАР – вода третього компоненту, в залежності від його природи, може або перешкоджати міцелоутворенню або сприяти цьому процесу. Введення, наприклад, низщих спиртів, але в малій кількості, приводить до зниження ККМ, тобто це полегшує міцелоутворення. Це явище – введення в склад міцел третього компоненту, який не розчиняється або слабко розчиняється в дисперсійному середовищі, називається солюбілізацією, або колоїдним розчиненням. Розрізняють пряму солюбілізацію (в водних дисперсіях ПАР) і зворотню (в карбоногідрогенних системах).
Солюбілізація широко застосовується в промисловості при виготовленні синтетичних латексів, для збільшення здобичі нафти, в емульсійній полімеризації, в поліграфічному, харчовому і фармацевтичному виробництвах.