

При врахуванні фазових перетворень застосовують рівняння у такому вигляді:

 (1.31)
(1.31)
Наприклад, розглядаючи процес утворення карбіду бору, що відбувається при температурі 1800 К за реакцією:
![]()
при якому плавляться магній (723 К) і бор оксид (923 К), випаровується магній (1390 К), тепловий ефект реакції за рівнянням виразиться як:
![]()
![]()
Другий закон термодинаміки. Ентропія. В природі всі фізичні, хімічні і біологічні перетворення здійснюються у певному напрямку і до певної межі. Усі ці перетворення відбуваються довільно. Вони є термодинамічно незворотні, так як завжди є втрати енергії системою в навколишнє середовище у вигляді тепла, тертя, випромінювання. Незворотні термодинамічні процеси можуть бути двох типів: несамовільні і самовільні. Для здійснення перших необхідно прикласти енергію ззовні, другі – відбуваються без витрати енергії зовні. Напрямок проходження самовільних процесів встановлює другий закон термодинаміки:
-
самовільно можуть відбуватися тільки ті процеси, при яких система переходить в найбільш імовірний стан;
-
теплота не може самовільно переходити від тіла з меншою температурою до тіла з більшою температурою;
-
різні види енергії прагнуть перейти в теплоту, а теплота прагне рівномірно розподілитись між всіма тілами.
Напрямок, в якому самовільно відбувається хімічна реакція, визначається сумісною дією двох факторів:
1) тенденцією до переходу системи в стан з найменшою внутрішньою енергією (для ізобарних процесів – з найменшою ентальпією);
2) тенденцією до досягнення імовірного стану (W), тобто стану, який може бути реалізований найбільшим числом рівномірних мікростанів.
Мірою імовірності стану системи в термодинаміці прийнято ентропію – S, величину пропорційну логарифму числа рівномірних мікростанів, якими може бути реалізований даний макростан:
![]() (1.32)
(1.32)
де
k
– стала Больцмана![]() ,
,
![]() - стала Авогадро.
- стала Авогадро.
Ентропія є мірою безпорядку в системі, мірою хаотичності розміщення частинок в речовині або тіл в системі. Вона характеризує ту частину енергії в незворотних процесах, яка не перетворюється в роботу, а розсіюється в навколишнє середовище у вигляді теплоти.
На величину ентропії речовин впливає їх природа і кількість (з ростом частинок ентропія зростає), а також умови, в яких находяться речовини (тиск, об'єм, температура, агрегатний стан).
Значення
абсолютної ентропії речовин розраховують
при стандартних умовах (р
= 101,325 кПа, Т
= 298 К,
t
= 250С)
і позначають
![]() (стандартна ентропія). Розмірність
(стандартна ентропія). Розмірність
![]() ─
─![]() .
.
Прийнято рахувати, що при 0К ентропія речовини дорівнює нулю, оскільки при цій температурі розміщення атомів або молекул в кристалічній структурі характеризується максимальним порядком.
З підвищенням температури ентропія завжди зростає. Вона стрибкоподібно збільшується при послідовному переході речовини з твердого кристалічного стану у газоподібний стан.
На рис.1.6 приведена типова крива зміни ентропії речовини при її нагріванні. Відрізок ОА характеризує поступове збільшення значень ентропії від 0 до температури плавлення (ТПЛ). В точці плавлення А відбувається стрибок ентропії (відрізок АВ) на ΔSПЛ. Відрізок ВС характеризує збільшення значень ентропії при нагріванні рідкої речовини від температури кипіння (ТКИП). В точці кипіння С також відбувається стрибок ентропії (відрізок СD) на ΔSВИП. Стрибкоподібна зміна ентропії в точках плавлення і кипіння відбувається при постійній температурі. При подальшому нагріванні (крива DE) ентропія газоподібної речовини знов плавно зростає.
|
|
|
Рис.1.6. Залежність ентропії від температури |
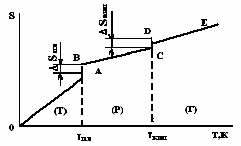
В разі збільшення тиску в системі або при охолодженні зміна ентропії відбувається в зворотному напрямку, яка характеризується кривими ЕD, СВ і АО. При цьому в точках D (початок конденсації) і В (початок кристалізації) спостерігається стрибкоподібне зменшення значень ентропії при сталих температурах. Зміна значень ентропії в процесах конденсації (ΔSКОНД) і кристалізації (ΔSКР) дорівнює значенням ΔSКИП і ΔSПЛ відповідно.
Ентропія як функція системи підкоряється закону Гесса. Це можна довести розглядаючи процеси переходу води із одного стану в інший. Так процес переходу льоду у пару можливий за двома шляхами.
Перший – через плавлення і випаровування рідини. В цьому випадку зміна ентропії складає:
![]()
Другий шлях – безпосередній перехід молекул води із льоду в пару – сублімація (возгонка). Зміна ентропії сублімації дорівнює:
![]() Отже,
зміна ентропії, як і зміна ентальпії,
не залежить від способу переходу, при
умові, що вихідний і кінцевий стани
речовини однакові. Це ілюструє рис.1.7
на якому показані зміни ентропії і
ентальпії води при фазових переходах.
Отже,
зміна ентропії, як і зміна ентальпії,
не залежить від способу переходу, при
умові, що вихідний і кінцевий стани
речовини однакові. Це ілюструє рис.1.7
на якому показані зміни ентропії і
ентальпії води при фазових переходах.
|
|
|
Рис.1.7. Зміна ентропії і ентальпії при фазових переходах |

З наведеного випливає, що зміна ентропії складного процесу, дорівнює сумі змін ентропії кожної окремої стадії цього процесу:
![]() (1.33)
(1.33)
Наприклад, якщо переохолоджений лід при температурі -100 С поступово нагрівати до перетворення його у пару з температурою 1200 С загальна зміна ентропії буде дорівнювати сумі змін ентропії таких стадій:
1) нагрівання льоду від –10 до 00 С, ΔS1;
2) плавлення льоду, ΔSПЛ;
3) нагрівання води від 0 до температури кипіння 1000 С, ΔS2;
4) випаровування води, ΔSВИП;
5) нагрівання пари від 100 до 1200 С, ΔS3.
За другим законом термодинаміки при нагріванні системи (абсолютна ентропія) чисельно дорівнює:
![]() (1.34)
(1.34)
де С - молярна теплоємність речовини.
Використовуючи значення стандартної ентропії визначають абсолютну ентропію при будь-якій температурі за рівнянням:
![]() (1.35)
(1.35)
Загальний вираз для розрахунку температурної залежності ентропії речовини, що знаходиться в стандартному стані і характеризується фазовими переходами, має вигляд:
![]()
 (1.36)
(1.36)
Для хімічної реакції, що відбувається при температурі Т, зміна ентропії хімічного процесу дорівнює різниці абсолютних ентропій продуктів реакції і вихідних речовин:
 (1.37)
(1.37)
Енергія Гібсса. Напрямок хімічної реакції. Узагальнені рівняння першого і другого законів термодинаміки (співвідношення Максвела) відображають зв'язок між ентропією і параметрами стану системи (тиск, температура, об'єм). Характеристичними функціями, виведеними на підставі співвідношень Максвела, є енергія Гіббса (ізобарно-ізотермічний потенціал):
![]() (1.38)
(1.38)
і енергія Гельмгольца (ізохорно-ізотермічний потенціал):
![]() (1.39)
(1.39)
Ізобарний і ізохорний потенціали є функціями стану системи і їх використовують для визначення напрямку процесу в умовах термодинамічної рівноваги.
Перебіг самочинного процесу в неізольованій системі можливий лише в напрямку, при якому за умов Т=const, Р=const зменшується енергія Гіббса і за умов Т=const, V=const зменшується енергія Гельмгольца, тобто ΔG < 0 і ΔF < 0.
Якщо ΔG > 0 і ΔF > 0 зміна стану системи відбувається тільки при використанні зовнішньої роботи. У випадку коли ΔG і ΔF дорівнюють нулю, то система перебуває у рівновазі.
Зміну енергії Гіббса в хімічній реакції розраховують за рівнянням Гіббса – Гельмгольца:
![]() (1.40)
(1.40)
В цьому рівнянні



В температурному інтервалі 298 – Т враховується температурна залежність ΔG˚Т = f(Т) у вигляді рівності:
![]() (1.41)
(1.41)
Рівняння (1.41) вирішується методом розкриття інтегралів:
![]()

Можливе виключення інтегральних величин Δс’/Т або Δb·Т+ Δс’/Т2 , що спрощує розрахунок, але при цьому точність розрахунку зменшується.
З рівняння Гіббса-Гельмгольца випливає:
1) якщо ΔН < 0 і ΔS > 0 то завжди ΔG < 0, тобто реакція з виділенням теплоти і збільшенням степені невпорядкованості можлива при всіх температурах;
2) якщо ΔH > 0 і ΔS < 0, то завжди ΔG > 0, тобто реакція з поглинанням теплоти і збільшенням степені упорядкованості неможлива ні при яких умовах;
3) у рештах випадках ( ΔН < 0, ΔS < 0 і ΔН >0, ΔS > 0) знак ΔG залежить від співвідношення членів ΔН і ТΔS. Реакція можлива, тільки якщо вона супроводжується зменшенням ізобарного потенціалу;
4) при кімнатній температурі, коли значення Т невелике, значення добутку ТΔS також невелике (рис.1.8), і звичайно зміна ентальпії переважує ТΔS. Тому більшість реакцій, які відбуваються при кімнатній температурі, - це реакції з виділенням теплоти (ΔН < 0). При збільшенні температури збільшується і добуток ТΔS, тому при високих температурах навіть реакції з поглинанням теплоти (ΔН > 0) стають самовільними.
|
|
|
Рис.1.8. Залежність ∆Н і ∆G від температури |

Слід відмітити, що хімічні реакції, під час перебігу яких відбувається зменшення термодинамічних потенціалів, називають екзергонічними, а якщо потенціали зростають – ендергонічними. При ізохорному процесі самовільна реакція можлива тільки при ΔF < 0. Для ΔF тлумачення точно такі, як і для ΔG. Чисельно ΔG і ΔF відрізняються на роботу розширення.
Зв'язок між основними термодинамічними функціями ілюструє діаграма, що зображена на рисунку:
|
|
|
Рис. 1.9. Діаграма співвідношень основних термодинамічних функцій |

З аналізу співвідношень витікає, що:
при Т→ 0 і Р → 0 ∆G → ∆F, а при Т = 0, Р = 0 і ∆n = 0 ∆G = ∆F.
Крім того, чим більше ∆n і тиск в реакційній системі, тим більше чисельне значення роботи розширення.
В табл. 1.1. співставлені форми характеристичних функцій, їх зміна при одному постійному параметрі стану і сумарні інтегральні форми.
Таблиця 1.1
Характеристичні функції і їх перемінні
|
Характеристичні функці |
Параметри стану |
Диференційна функція |
Прийнята інтегральна форма |
|
Внутрішня енергія U (ізохорно-ізоентропійний потенціал) |
S, V |
dV-TdS-PdV |
При S=const ΔUS = -RTln(V2/V1) При V=const ΔUV=T(S2-S1) U=F+TS |
|
Ентальпія Н (ізобарно-ізоентропійний потенціал) |
S, P |
dH-TΔS+VdP |
При S=const ΔНS = RTln(Р2/Р1) При Р=const ΔНР=T(S2-S1) Н=U+pV |
продовження табл. 1.1
|
Енергія Гельмгольца F (ізохорно-ізотермічний потенціал) |
V, T |
dF=-PdV-SdT |
При V=const ΔFV = Qутв.ln(T1/T2) При T=const FT=ΔUT F=U-TS |
|
Енергія Гіббса (ізобарно-ізотермічний потенціал) |
P, T |
dG=VdP-SdT |
При P=const ΔGP = Qутв.ln(T1/T2) При T=const GT= RTln(Р2/Р1) G=F+pV |
Теплота фазових перетворень. Залежність теплоти фазового перетворення від умов його проходження для будь-якого рівноважного процесу визначається за рівнянням Клаузіуса – Клапейрона:
![]() (1.42)
(1.42)
де НФ.П. – теплота фазового перетворення (плавлення, випаровування, поліморфного перетворення, сублімації тощо); ∆V – зміна об'єму при фазовому переході, dp/dT – похідна, що зв'язує зміну температури і тиску за умови збереження стану рівноваги між фазами.
Теплота плавлення – переходу твердої фази в рідку - завжди позитивна. Об'єм рідкої фази (VР) в загальному випадку може бути більше або менше об'єму в тій же кількості твердої фази (VТ). Звідси витікає, що величина dp/dТ, яка характеризує зміну температури від тиску, може бути позитивною або від'ємною тобто, температура плавлення може підвищуватись або знижуватись зі збільшенням тиску.
Величина dТ/dР має від'ємне значення лише для води і деяких інших речовин, у яких густина рідини при температурі плавлення більше густини твердої фази.
Рівняння Клаузіуса-Клапейрона в цьому випадку набуває вигляду:
![]() (1.43)
(1.43)
При випаровуванні – переходу рідкої фази в газоподібну – теплота випаровування також позитивна, а температура випаровування завжди підвищується зі зростанням тиску.
В зв'язку з тим, що мольний об'єм пари значно перевищує мольний об'єм рідини значення Vр в рівнянні можна знехтувати і воно прийме вигляд:
![]() (1.44)
(1.44)
Якщо насичену пару рахувати ідеальним газом, тобто
![]()
Тоді рівняння (1.44) набуде вигляду:
![]() (1.45)
(1.45)
В області невисоких тисків теплоти випаровування порівняно мало змінюється з температурою, тому її можна вважати сталою. В цьому випадку після перетворення цього рівняння дістанемо рівняння:
|
|
або |

![]() (1.46)
(1.46)
які широко застосовуються в практиці.
Практичне застосування термодинамічних розрахунків. Розглянемо розрахунок основних термодинамічних величин на прикладі хімічної реакції, яка відбувається в гомогенній системі:
|
Н2(Т)+СО2(Г)=Н2О(Г)+СО(Г); |
∆НТ=? |
при температурі 2000 К і тиску 3 атмосфери.
З довідника виписуємо термодинамічні величини для вихідних сполук і продуктів реакції.
|
Речовина |
∆Н0298, кДж/моль |
S0298, Дж/(мольК) |
Теплоємність, Дж/(моль К) |
Темпера-турний інтервал, К |
||
|
Коефіцієнти рівняння Ср=f(T) |
||||||
|
a |
b |
c |
||||
|
Н2(г) |
0 |
130,6 |
27,28 |
3,26 |
0,50 |
298 – 3000 |
|
СО2(г) |
- 393,5 |
213,6 |
44,14 |
9,04 |
- 8,53 |
298 – 2500 |
|
Н2О(г) |
- 241,8 |
188,7 |
30,00 |
10,71 |
0,33 |
298 – 2500 |
|
СО(г) |
- 110,5 |
197,4 |
20,41 |
4,10 |
- 0,46 |
298 – 5000 |
За рівнянням Гесса визначаємо зміну стандартної ентальпії:

![]()
![]()
оскільки
![]() реакція ендотермічна.
реакція ендотермічна.
Стандартну зміну ентропії визначаємо за рівнянням:

![]()
![]()
Зміну стандартного потенціалу
![]() (енергія Гіббса) визначаємо за рівнянням
Гіббса-Гельмгольца за першого припущення
(∆Ср = 0):
(енергія Гіббса) визначаємо за рівнянням
Гіббса-Гельмгольца за першого припущення
(∆Ср = 0):
![]()
Оскільки
![]() реакція
необоротна.
реакція
необоротна.
Залежність теплоємності всіх речовин (газів), що приймають участь в реакції, від температури визначаємо за температурним рядом:
![]()
Тоді зміна теплоємності для хімічної реакції буде:

![]()

За стандартних умов:
![]() При
другому наближенні (СР=const)
приймаємо:
При
другому наближенні (СР=const)
приймаємо:
![]()
тоді залежність ентальпії від температури (1.28) буде мати вигляд:

Значення ентальпії при температурі реакції складе:
![]()
Аналогічно знаходимо ентропію реакції, користуючись рівнянням:

![]()
Більш
точний розрахунок значень ентальпії і
ентропії проведемо, застосовуючи
температурну залежність теплоємності
![]() хімічної реакції:
хімічної реакції:
![]()




За ентропійною формулою, за умови ∆Ср = const, значення енергії Гіббса при температурі реакції дорівнює:
![]()
а враховуючи температурну залежність ∆Ср = f (T) складає:
![]()
В першому наближенні (Ср = 0, ∆Н0Т = ∆Н0298 = 41200 Дж) за рівнянням Гіббса-Гельмгольца (у формі невизначеного інтеграла):
![]()
знаходимо значення постійної інтегрування В для Т = 298 К:
![]()
![]()
Тоді:
![]()
В другому наближенні (∆Ср = const = ∆С0р 298):
![]()
Звідки постійна інтегрування В при Т = 298 К дорівнює:
![]()
![]()
Тоді,
![]()
При більш точному розрахунку значення ∆G0T:
![]() маємо:
маємо:![]()

або
 .
.
звідки В = -137,2.
Тоді:

Розглянемо термодинамічний розрахунок для хімічної реакції, що відбувається в гетерогенній системі при температурі 500–700 К.
Вихідні дані для реакції:
![]()
|
Речовина |
∆Н0298, кДж/моль· К |
S0298, кДж/мол·ьК |
Теплоємність, Дж/(мольК) |
Темпера-турний інтер- вал, К |
||
|
Коефіцієнти рівняння Ср = f(T) |
||||||
|
a |
b |
c |
||||
|
MnO2(K) |
-519,65 |
53,14 |
69,45 |
10,21 |
-16,23 |
273 –773 |
|
HCl(Г) |
-92,30 |
186,70 |
26,53 |
4,60 |
1,09 |
298 –2000 |
|
MnCl2(K) |
-468,61 |
117,15 |
75,48 |
13,22 |
-5,73 |
273 –923 |
|
H2O(Г) |
-241,84 |
188,74 |
30,00 |
10,71 |
0,33 |
298–2500 |
|
Cl2(Г) |
0 |
223,0 |
36,69 |
1,05 |
-2,52 |
273-1500 |
Визначаємо зміну стандартних ∆Н0298 і ∆S0298:

(реакція екзотермічна).
 .
.
Зміну енергії Гіббса ∆G0298 визначаємо за ентропійним рівнянням при першому припущенні (∆Ср = 0):
![]()
Оскільки
![]() реакція при температурі 298 К
відбувається самовільно.
реакція при температурі 298 К
відбувається самовільно.
Зміну теплоємності для хімічної реакції знаходимо за температурними рядами:
![]()


Приймаючи ∆Ср=∆С0р298 знаходимо зміну ентальпії та ентропії системи:
![]()

При більш точному розрахунку (∆С р= f(Т)) ці рівняння мають вигляд:
![]()
![]()

![]()

За наближеними і більш точними рівняннями розраховуємо значення ∆Н0Т та ∆S0Т при температурах реакції 500 і 700 К:
![]()
![]()
![]()
![]()


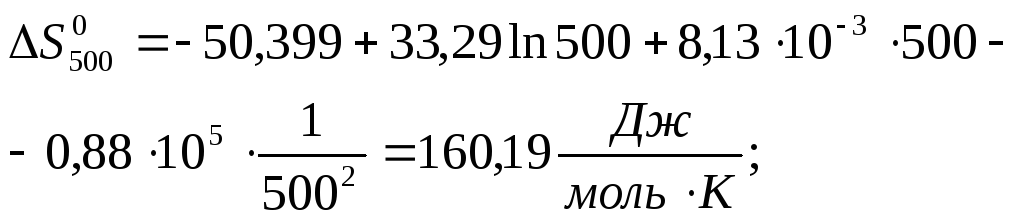

Розрахунок ∆G0T проводимо за ентропійними формулами при припущенні, що ∆Ср = const:
![]()
![]()
При припущені, що ∆Ср = f (T) ∆G0T дорівнює:
![]()
![]()
За
рівнянням Гіббса-Гельмгольца при першому
наближенні (∆Ср
= 0,
![]() Дж),
використовуючи значення
Дж),
використовуючи значення
![]() Дж
при температурі
298 К
знаходимо постійну інтегрування В:
Дж
при температурі
298 К
знаходимо постійну інтегрування В:
![]()
![]()
Тоді:
![]()
![]()
При другому наближенні (∆Ср = const = ∆Cp298):
![]()

Для Т = 298 К:
![]()
Звідки В = 111,7
Тоді:![]()
![]()
При більш точному розрахунку (Ср = f (T)):
![]()

Для Т = 298 К:
![]()

Звідки В = 82,69.
Тоді:


Оцінка
похибки при розрахунках
![]() ,
,
![]() і
і
![]() виконується
в порівнянні з найбільш точним результатом.
Для ∆
виконується
в порівнянні з найбільш точним результатом.
Для ∆![]() при температурі 500 К
різниця між точним і наближеним значенням
складає:
при температурі 500 К
різниця між точним і наближеним значенням
складає:
55825,6 – 85822,8=2,8 (Дж), або
відносна похибка:
![]()
Для
![]() :
:
![]()
і
![]()
![]()