

Запитання для самоконтролю
1. Чи залежить величина радіуса йонної атмосфери від концентрації йонів, їх природи, природи розчинника? Зробіть оцінку радіуса йонної атмосфери для 0,001 і 0,01 молярних розчинів 1,1- заряджених електролітів у воді згідно 1 наближенню теорії Дебая-Гюккеля.
2. Як відрізняють значення коефіцієнтів активності водних розчинів: а) КС1 і NaC12, б) КС1 і ВаС12, в) ВаС12 і BaSO4, якщо розчини однакової йонної сили (І випадок) і однакової молярної концентрації (ІІ випадок)?
3. Чому значення коефіцієнтів активності розчинів: КС1 у воді і КС1 в метиловому спирті відрізняються між собою? Відповідь підтвердити розрахунком.
4.
Для 0,001 молярного водного розчину
електролітів: NaCl,
BaCl2,
Na2SO4,
ZnSO4
розрахувати значення коефіцієнтів
активності
![]() за рівнянням теорії Дебая-Гюккеля і
порівняти їх з довідковими.
за рівнянням теорії Дебая-Гюккеля і
порівняти їх з довідковими.
5. Обчислити коефіцієнти активності та активності йонів 0,01 М розчину Al2(SO4)3.
6. Як зміниться коефіцієнт активності 0,01 молярного водного розчину NaCl, якщо до нього добавили 0,015 моль/л Fe2(SO4)3 (T = 298 К)?
7. Визначити концентрацію йонів гідрогену і рН 0,5 %-го розчину сульфатної кислоти.
8. Як зміниться розчинність AgBrO3 в 0,02 молярному водному розчині AgNO3 порівняно з його розчинністю у воді (S = 0,0018 моль/л)?
Тема 3. Хімічна кінетика
Класифікація хімічних реакцій. Усі хімічні реакції проходять або в гомогенних системах, що складаються лише з однієї фази, або в гетерогенних, які складаються з двох і більше фаз. Такі реакції називаються відповідно гомогенними і гетерогенними.
В гетерогенних системах одна із фаз завжди перебуває в диспергованому стані, тому хімічна взаємодія в них відбувається на поверхні поділу фаз. Розрізняють двофазні системи „рідина-тверда фаза”, „рідина-рідина” (що взаємонерозчинні), „газ-рідина”, “газ-тверда речовина”, „тверда речовина – тверда речовина”.
Гетерогенні реакції відбуваються досить повільно ніж гомогенні. Це обумовлено складністю їх механізму: дифузія до поверхні поділу фаз, хімічна взаємодія реагентів, дифузія продуктів реакції від поверхні поділу фаз, поверхневі явища та ін.
За механізмом усі реакції можна поділити на дві групи: прості і складні. Прості реакції складаються із однієї стадії – хімічного перетворення. За ознакою молекулярності вони поділяються на одномолекулярні (мономолекулярні), двомолекулярні (бімолекулярні), тримолекулярні.
До складних реакцій відносяться ланцюгові, спряжені, паралельні, послідовні, оборотні, фотохімічні радіаційно-хімічні, ферментативні, гомогенно-гетерогенні. Для складних реакцій молекулярність можна визначити тільки для їх окремих стадій.
Усі хімічні реакції формально поділяють на реакції першого, другого і третього порядків, а також нульового і дробного порядків. Під порядком реакції розуміють суму показників ступенів при змінюючих концентраціях реагентів в кінетичному рівнянні. Суму стехіометричних коефіцієнтів вихідних речовин, що одночасно беруть участь в елементарному акті реакції, прийнято визначати як її молекулярність.
На швидкість гомогенних гетерогенних реакцій впливають: природа реагуючих речовин, концентрація речовин, тиск, температура, наявність каталізатора (ферментів). У випадку гетерогенних реакцій на їх швидкість також впливає ступінь диспергованості (поверхня поділу фаз) речовин.
Знаючи хімічну кінетику реакцій можна визначати оптимальні параметри і умови проведення різних хіміко-технологічних і біохімічних процесів, які відбуваються при виробництві хімічних матеріалів, синтезу біологічних речовин, нейтралізації і знешкодження шкідливих речовин, забруднювачів навколишнього середовища.
Швидкість хімічної реакції. Кінетичні закономірності перебігу хімічних реакцій ґрунтуються на припущенні про те, що реагують тільки ті молекули, які стикаються між собою. Кількість таких зіткнень прямо пропорційна кількості молекул, тому швидкість реакції повинна бути пропорційною концентрації реагуючих речовин (закон діючих мас).
Для емпіричного рівняння:
![]()
Швидкість прямої реакції можна записати як:
![]() (3.1)
(3.1)
де
v
- швидкість хімічної реакції;
![]() і
і
![]() - концентрації реагуючих речовин; а
і b
- їх стехіометричні коефіцієнти. Правило,
виражене рівнянням, називають основним
постулатом хімічної кінетики.
- концентрації реагуючих речовин; а
і b
- їх стехіометричні коефіцієнти. Правило,
виражене рівнянням, називають основним
постулатом хімічної кінетики.
З рівняння випливає, що швидкість реакції, будучи функцією концентрації, залежить також від часу, оскільки концентрація реагуючих речовин з часом змінюється.
|
|
|
Рис.3.1.
Графічна залежність зміни концентрації
D(2)
з часом
|

На рисунку 3.1 приведені графічні залежності зміни концентрації вихідного реагенту (крива 1) і продукту реакції (крива 2) з часом, а також середня і істина швидкості хімічної реакції.
|
|
|
Рис.3.2.
Середня і істина швидкості хімічної
реакції
|

Для
заданого інтервалу часу
![]() можна ввести поняття про середню
швидкість реакції
можна ввести поняття про середню
швидкість реакції
![]() (рис.3.2):
(рис.3.2):
![]() (3.2)
(3.2)
де
![]() і
і
![]() - концентрація реагенту А
в початковій
- концентрація реагенту А
в початковій
![]() і кінцевий
і кінцевий
![]() момент часу.
момент часу.
За умови, що
![]()
![]()
швидкість хімічної реакції в даний момент часу відповідає значенню миттєвої (істинної) швидкості реакції v:
![]() (3.3)
(3.3)
Для реагенту А середня швидкість реакції виразиться як:
![]() (3.4)
(3.4)
В той же час, для продукту реакції D швидкість реакції буде мати знак плюс:
![]() (3.5)
(3.5)
тому, що його концентрація збільшується.
Швидкість
реакції в даний момент часу можна
визначити графічним способом. Для цього
до точки на кривій зміни концентрації
реагенту А,
яка відповідає даному моменту часу
(приклад
![]() )
проводять дотичну. Тангенс кута нахилу
)
проводять дотичну. Тангенс кута нахилу
![]() дотичної до кривої відповідає швидкості
реакції.
дотичної до кривої відповідає швидкості
реакції.
Отже, хімічна кінетика визначає поняття швидкості гомогенної реакції як зміну концентрації одного з реагуючих компонентів за одиницю часу в одиниці об’єму (при постійній температурі):
![]() (3.6)
(3.6)
де
![]() - об’єм реакційного елемента.
- об’єм реакційного елемента.
Гетерогенні
реакції відбуваються на поверхні
![]() поділу фаз (каталізатора), яка в момент
взаємодії реагентів практично не
змінюється. Тому швидкість гетерогенних
реакцій описується рівнянням:
поділу фаз (каталізатора), яка в момент
взаємодії реагентів практично не
змінюється. Тому швидкість гетерогенних
реакцій описується рівнянням:
![]()
У
рівнянні швидкості прямої реакції
коефіцієнт пропорційності
![]() не залежить від концентрації реагуючих
речовин. Його фізичний зміст можна
знайти, якщо прийняти, що концентрації
реагуючих речовин А
і В
дорівнюють одиниці, тобто
не залежить від концентрації реагуючих
речовин. Його фізичний зміст можна
знайти, якщо прийняти, що концентрації
реагуючих речовин А
і В
дорівнюють одиниці, тобто
![]()
За цих умов:
![]()
Це
і є фізичний зміст коефіцієнта
![]() .
Оскільки константа швидкості реакції
не змінюється і за сталих умов (природа
реагуючих речовин, тиск, температура,
каталізатор) є величиною постійною,
вона показує з якою швидкістю відбувається
ця реакція, якщо концентрації реагуючих
речовин сталі і дорівнюють одиниці.
.
Оскільки константа швидкості реакції
не змінюється і за сталих умов (природа
реагуючих речовин, тиск, температура,
каталізатор) є величиною постійною,
вона показує з якою швидкістю відбувається
ця реакція, якщо концентрації реагуючих
речовин сталі і дорівнюють одиниці.
На константу швидкості впливає в основному температура. Фактор впливу температури на константу швидкості гомогенної реакції виражає рівняння Арреніуса:
![]() (3.7)
(3.7)
де А і В – характерні для даної реакції сталі, що не залежать від температури.
|
|
|
Рис. 3.3. Графічна залежність логарифма константи швидкості хімічної реакції від оберненої температури |

Рівняння Арреніуса в диференціальній формі має вигляд:
![]() (3.8)
(3.8)
Величина Е називається арреніусовською енергією активації. Її розглядають як енергетичний бар’єр, що подолають лише активні молекули.
Значення енергії активації дістають з експериментальних даних. Для цього будують графічну залежність lnk і 1/T в прямокутній системі координат.
Ця залежність є прямою лінією, за кутом нахилу якої визначають енергію активації реакції:
![]()
де m - відношення масштабу по осі абсцис до масштабу по осі ординат.
Відрізок, що відсікається продовженням прямої на осі ординат, відповідає ln/k0.
Знаючи константу швидкості при двох значеннях температури, можна розрахувати енергію активації:
![]() (3.9)
(3.9)
Енергію активації можна визначити і за виразом:
 (3.10)
(3.10)
Вплив температури і енергії активації на швидкість хімічних реакцій можна виразити рівнянням Арреніуса в експоненціальному вигляді:
![]() (3.11)
(3.11)
де
![]() - передекспоненціальний множник,
пропорційний числу зіткнень молекул.
- передекспоненціальний множник,
пропорційний числу зіткнень молекул.
Якщо концентрації реагуючих речовин дорівнюють 1 моль/л, то рівняння Арреніуса дає змогу виразити залежність швидкості реакції від температури:
![]() (3.12)
(3.12)
Оскільки в рівнянні температура входить у показник ступеня, то швидкість хімічних реакцій значною мірою залежить від зміни температури.
Згідно з емпіричним правилом Вант-Гоффа: підвищення температури на кожні 10 градусів збільшує швидкість реакції приблизно в 2 - 4 рази.
У математичній формі правило Вант-Гоффа записується так:
![]() (3.13)
(3.13)
де
![]() - збільшення температури;
- збільшення температури;
![]() і
і
![]() - швидкість реакції до
- швидкість реакції до
![]() і після підвищення температури до
і після підвищення температури до
![]() ;
;
![]() - температурний коефіцієнт швидкості
реакції
- температурний коефіцієнт швидкості
реакції
![]() .
.
Температурний коефіцієнт можна знайти за виразом:
![]() (3.14)
(3.14)
Рівняння Вант-Гоффа є приблизним, оскільки швидкість реакції, крім температури залежить також від енергії активації, яка в свою чергу, залежить від температури.
Молекулярність і порядок хімічних реакцій. Молекулярніть простої реакції можна пов’язати із стехіометричним рівнянням. Наприклад, реакція розкладання пероксиду гідрогену:
![]()
є мономолекулярною, а реакція синтезу хлориду гідрогену:
![]()
бімолекулярною.
Прикладом тримолекулярної реакції є реакція синтезу води з Гідрогену і Оксигену:
![]()
Найчастіше реакції бувають бімолекулярними, тримолекулярні реакції трапляються рідко, а більш молекулярні взагалі невідомі.
Для елементарних реакцій порядок, як правило, співпадає з їхньою молекулярністю. Складні реакції здійснюються через проміжні стадії, які забезпечують найбільшу швидкість реакції. Для таких реакцій порядок не співпадає з коефіцієнтами сумарного стехіометричного рівняння. Наприклад, мономолекулярна реакція розкладання пероксиду гідрогену може відбуватися, як бімолекулярна:
![]()
Порядок цієї реакції повинен бути другим. Проте насправді, залежно від умов, порядок цієї реакції є першим або дробовим.
У тих випадках, коли за умовами експерименту різниця між концентраціями вихідних речовин дуже велика порядок реакції також не співпадає з її молекулярністю.
Порядок реакції на практиці визначають методом ізоляції Оствальда. Згідно цього метода усі реагенти, крім одного, беруть з надлишком (їх концентрація практично стала в ході реакції) і тому їх вводять в константу швидкості. Тоді, для реагента А концентрація, якого змінюється, вираз швидкості реакції буде мати вигляд:
![]() (3.15)
(3.15)
Рівняння логарифмують:
![]() (3.16)
(3.16)
і
будують графічну залежність
![]() від
від
![]() .
Тангенс кута нахилу прямої цієї залежності
відповідає
.
Тангенс кута нахилу прямої цієї залежності
відповідає
![]() .
Аналогічно визначають порядки реакції
за всіма іншими реагентами.
.
Аналогічно визначають порядки реакції
за всіма іншими реагентами.
|
|
|
Рис. 3.4. Залежність логарифма швидкості від логарифма концентрації вихідної речовини |

Реакції нульового порядку. Гомогенних хімічних реакцій нульового порядку, в принципі, не існує. Але, якщо речовина, за якою контролюється хід реакції, не бере участі в лімітуючій стадії складного процесу швидкість реакції не залежить від концентрації реагенту і графічно виражається прямою лінією (див.рисунок). Тип таких реакцій називається реакціями нульового порядку:
![]() (3.17)
(3.17)
де
![]() - константа швидкості реакції нульового
порядку.
- константа швидкості реакції нульового
порядку.

Нульовий порядок спостерігається в гетерогенних реакціях в яких швидкість витрачання речовини значно менша від її надходження. До реакції нульового порядку також належать процеси ферментативного каталізу.
Поєднуючи
рівняння
![]() і
і
![]() маємо:
маємо:
![]() (3.18)
(3.18)
Інтегрування цього рівняння призводить до рівняння:
![]() (3.19)
(3.19)
Яке дає змогу визначити константу швидкості реакцій нульового порядку. Кінетичне рівняння хімічної реакції нульового порядку має вигляд:
![]() (3.20)
(3.20)
Експериментально
константу швидкості можна знайти,
користуючись графічною залежністю
![]() від
від
![]() .
Тангенс кута нахилу прямої лінії дорівнює
константі швидкості
.
Тангенс кута нахилу прямої лінії дорівнює
константі швидкості
![]() .
.
За кінетичним рівнянням хімічної реакції нульового порядку константа швидкості реакції залежить від способу вираження концентрації вихідної речовини:
![]() моль/(л. с).
моль/(л. с).
Користуючись
значеннями величини константи швидкості,
можна розрахувати час закінчення реакції
![]() :
:
![]()
|
|
|
Рис.3.5.
Залежність
|

Для визначення періоду напівперетворення – час, за який концентрація вихідних речовин зменшується в два рази, застосовуємо рівняння:
|
при |
|
|
(3.21) |
Кількість
речовини х,
що прореагувала за час
![]() ,
можна розрахувати, якщо прийняти:
,
можна розрахувати, якщо прийняти:
|
|
і |
|
і ввести у кінетичне рівняння реакції:
![]()
де
а
– початкова концентрація реагенту при
![]() ;
х
– кількість речовини, що прореагувала
за час
;
х
– кількість речовини, що прореагувала
за час
![]() ;
;
![]() - об’єм суміші, яка реагує.
- об’єм суміші, яка реагує.
При
![]() :
:
![]()
Реакції першого порядку. До них належать реакції ізомеризації, термічного розкладання речовин, радіоактивного розпаду атомних ядер, багато бімолекулярних реакцій при умові, що концентрація одної із речовин, що реагує, підтримується постійною.
Для необоротної реакції першого порядку типу:
![]()
швидкість реакції у диференціальній формі запишемо так:
![]() (3.22)
(3.22)
![]() (3.23)
(3.23)
в інтегральній формі:
![]() (3.24)
(3.24)
![]() (3.25)
(3.25)
в експоненціальній формі:
![]() (3.26)
(3.26)
де
![]() - початкова концентрація речовини А.
- початкова концентрація речовини А.
На
рисунку 3.6 приведена залежність
![]() для двох різних значень початкової
концентрації
для двох різних значень початкової
концентрації
![]() і
і
![]() .
Дотичні до кривих залежності, як бачимо,
перетинаються на абсцисі в одній точці
.
Дотичні до кривих залежності, як бачимо,
перетинаються на абсцисі в одній точці
![]() ,
величина якої не залежить від початкової
концентрації речовини А.
Час
,
величина якої не залежить від початкової
концентрації речовини А.
Час
![]() відповідає початковій швидкості
відповідає початковій швидкості
![]() :
:
![]()
звідки:
![]()
Рівняння в інтегральній формі дає можливість визначити константу швидкості необоротної реакції першого порядку:
|
|
або |
|
Величина k не залежить від способу вираження концентрації, її розмірність становить с-1 або хв.-1.
За
рівнянням 3.25 залежність
![]() від
від
![]() є лінійною з кутом нахилу, що дорівнює
– k.
Для різних значень
є лінійною з кутом нахилу, що дорівнює
– k.
Для різних значень
![]() ,
у напівлогарифмічних координатах
матимемо паралельні прямі (рис.3.7).
,
у напівлогарифмічних координатах
матимемо паралельні прямі (рис.3.7).
Згідно
з цим рівнянням розв’язання двох рівнянь
з різними значеннями
![]() дає:
дає:
![]()
|
|
|
Рис.
3.6. Дотичні до кривих залежності
|

|
|
|
Рис.3.7. Залежність логарифму концентрації компоненти А від часу |

Що дозволяє визначити k без знаходження початкової концентрації речовини. Для оцінки k можна скористатися любою іншою величиною, якщо вона пропорційна концентрації, наприклад, електропровідністю, оптичною густиною та ін.
Період напівперетворення для необоротної реакції першого порядку не залежить від початкової концентрації реагуючих речовин і дорівнює:
![]() (3.28)
(3.28)
Кількість
речовини х,
що прореагувала за час
![]() розраховують за рівнянням:
розраховують за рівнянням:
![]() (3.29)
(3.29)
де а – концентрація на початку реакції.
Якщо
замість концентрації скористатися
ступенем перетворення
![]() ,
то кінетичне рівняння матиме вигляд:
,
то кінетичне рівняння матиме вигляд:
![]()
Кінетика
обротних реакцій ускладнюється одночасним
протіканням оборотної реакції. Прикладом
оборотних реакцій є синтез амоніаку,
окиснення оксиду сульфуру(IV),
реакції рацемізації і ізомеризації, в
тому числі оптичної ізомеризації,
наприклад ізомеризації
![]() -глюкози
в
-глюкози
в
![]() -глюкозу.
-глюкозу.
Для оборотної реакції, що відбувається при сталому об’ємі за стехіометричним рівнянням:
![]()
справджується така залежність:
![]() (3.30)
(3.30)
При
досягненні рівноважного стану константа
хімічної рівноваги
![]() буде дорівнювати:
буде дорівнювати:

де
![]() і
і
![]() - концентрація речовин в стані рівноваги.
- концентрація речовин в стані рівноваги.
Враховуючи, що
|
|
і |
|
|
|
рівняння (3.30) після перетворень матиме вигляд:
![]() (3.31)
(3.31)
в інтегральній формі:
|
|
|
(3.32) |
|
або |
|
|

Початкова швидкість оборотної реакції становитиме:
![]() (3.33)
(3.33)
З цього рівняння випливає, що
![]()
На
рисунку 3.8 залежності
![]() для оборотної реакції першого порядку
для оборотної реакції першого порядку
![]() є проекцією на вісь абсцис точки перетину
дотичної до початкової ділянки кривої
при
є проекцією на вісь абсцис точки перетину
дотичної до початкової ділянки кривої
при
![]() для
для
![]() і рівноважної концентрації
і рівноважної концентрації
![]() .
.
|
|
|
Рис.3.8.
Залежність
оборотної реакції першого порядку |
Дотичні
для
![]() і
і
![]() перетинають абсцису в точці
перетинають абсцису в точці
![]() ,
величина якої не залежить від початкової
концентрації речовини А.
В цьому випадку швидкість реакції
становитиме:
,
величина якої не залежить від початкової
концентрації речовини А.
В цьому випадку швидкість реакції
становитиме:
![]() (3.34)
(3.34)
Час
![]() дорівнює:
дорівнює:
![]()
оскільки
на початку процесу добуток
![]() дорівнює нулю.
дорівнює нулю.
У
випадку, коли
![]() маємо:
маємо:
![]()
тоді
|
|
і |
|
Реакції
другого порядку.
Реакцією другого порядку називають
реакцію, швидкість якої пропорційна
або добутку концентрацій двох реагуючих
речовин (![]() і
і
![]() ),
або квадрату концентрацій одної із
реагуючих речовин (
),
або квадрату концентрацій одної із
реагуючих речовин (![]() ).
).
До них належать реакції розкладання, обміну і сполучення:
![]()
![]()
![]()
Так, реакція розкладання пероксиду гідрогену, яка вже згадувалась, при більш високих температурах відбувається не по першому порядку, а по другому:
![]()
Для
такого типу необоротних реакцій другого
порядку рівняння швидкості (при
![]() )
матиме вигляд:
)
матиме вигляд:
![]() (3.35)
(3.35)
Коефіцієнт 2 в рівнянні вказує на те, що в кожному елементарному акті хімічної взаємодії беруть участь дві молекули речовини А.
Здійснюючи
поділ змінних і, інтегруючи від
![]() до
до
![]() та від 0 до
та від 0 до
![]() дістанемо:
дістанемо:
 (3.36)
(3.36)
звідки:
|
|
і |
|

 (3.37)
(3.37)
Розмірність
![]() ,
тобто числове значення k
залежить від способу вираження
концентрації:
,
тобто числове значення k
залежить від способу вираження
концентрації:
![]() .
.
Відповідно з рівняння 3.36:
![]()
лінійна
залежність в координатах
![]() від
від
![]() свідчить про те, що реакція, яка
розглядається, має другий порядок
(рис.3.9), а тангенс кута нахилу
свідчить про те, що реакція, яка
розглядається, має другий порядок
(рис.3.9), а тангенс кута нахилу
![]() прямої дорівнює числовому значенню
константи швидкості k.
прямої дорівнює числовому значенню
константи швидкості k.
Період напівперетворення речовини А обернено пропорційний початковій концентрації реагуючих речовин:
![]() (3.38)
(3.38)
На рисунку 3.9 періоди напівперетворень зображені точками перетину прямої залежності з абсцисою.
Щоб
визначити кількість речовини х,
яка прореагувала за проміжок часу
![]() ,
враховуючи, що
,
враховуючи, що
![]() і
і
![]()
з рівняння 3.37 одержимо:
![]() (3.39)
(3.39)
де
-
![]() - замість 2k;
а
– маса реагуючої речовини в початковій
момент.
- замість 2k;
а
– маса реагуючої речовини в початковій
момент.
|
|
|
Рис.3.9.
Залежність
реакції другого порядку (2А=В+D) |

Звідси:
![]() (3.40)
(3.40)
Якщо в реакції кількість реагуючих речовин неоднакова константу швидкості реакції другого порядку знаходять інтегруючи такий вираз:
![]()
де
а
і b
– початкові концентрації першої і
другої речовини, х
– концентрації першої і другої речовини,
які прореагували за час
![]() .
.
Інтегрування цього рівняння дає вираз:
![]()
|
або |
|
(3.41) |
Для оборотної бімолекулярної реакції другого порядку типу:
![]()
рівняння її швидкості має вигляд:
![]() .
(3.42)
.
(3.42)
Значення швидкості оборотної реакції залежить від співвідношення реагентів.
У випадку коли:
![]()
швидкість утворення продуктів реакції виражається рівнянням:
![]() (3.43)
(3.43)
При досягненні рівноваги, враховуючи, що
![]()
маємо:
 (3.44)
(3.44)
Перетворюючі два останні рівняння одержуємо:

Проінтегрувавши це рівняння швидкості, дістанемо:
 (3.45)
(3.45)
|
де |
|
|


Графічна
залежність виразу А2
від часу
![]() (рис. 3.10) є прямою лінією з кутовим
коефіцієнтом, що дорівнює множнику біля
(рис. 3.10) є прямою лінією з кутовим
коефіцієнтом, що дорівнює множнику біля
![]() в рівнянні. Пряма відсікає від ординати
відрізок, що дорівнює значенню виразу
в рівнянні. Пряма відсікає від ординати
відрізок, що дорівнює значенню виразу
![]() в рівнянні.
в рівнянні.
Константи
швидкостей оборотної реакції цього
типу можна оцінити, як і оборотну реакцію
першого порядку за кутовим коефіцієнтом
дотичної на початковій ділянці кривої
![]() (див.вище).
(див.вище).
З виразу початкової швидкості оборотної реакції другого порядку:
![]()
|
|
|
Рис.3.10.
Залежність
другого
порядку (типу
|

при
![]() дістанемо
дістанемо
![]() :
:
![]()
а
![]() з виразу:
з виразу:
![]()
![]()
При
![]()
інтегрування загального рівняння швидкості оборотної реакції другого порядку дає:
![]() (3.46)
(3.46)
де
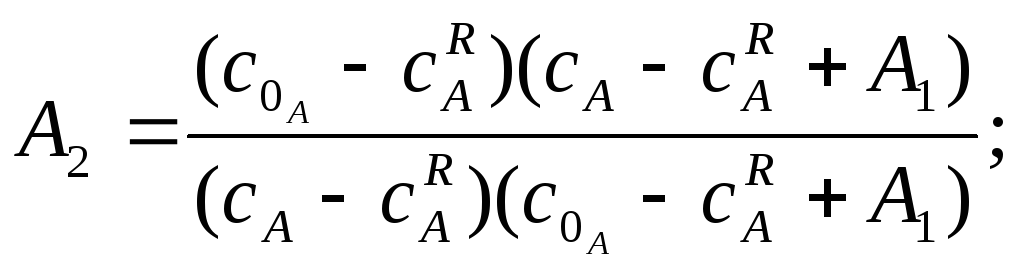
![]()
Реакції третього і n-го порядків. Швидкість необоротної хімічної реакції третього порядку, коли речовини А, В і D взяті в нееквівалентних кількостях (кількість молів a, b і d неоднакова) описується рівнянням:
![]()
Інтегрування цього рівняння дозволяє отримати вираз для розрахунку часу реакції за умов:
|
|
при |
|
 (3.47)
(3.47)
Якщо речовини В і D мають неоднакову кількість (b) молей, то швидкість описується рівнянням:
![]()
Відповідно час хімічної взаємодії можна розрахувати за формулою:
![]() (3.48)
(3.48)
На практиці порядки реакцій визначають користуючись методом Вант-Гоффа за рівняннями:
![]() і
і
![]()
Кінетичний
порядок може приймати також дробні і
навіть від’ємні значення. Так, швидкість
термічного розкладання ацетатного
альдегіду в газовій фазі пропорційна
його концентрації в ступені
![]() :
:
![]()
![]()
звідки
![]() .
.
В таблиці „Кінетичні закономірності для реакцій з різними кінетичними порядками” приведені кінетичні закономірності для реакцій з різними кінетичними порядками (див. додаток).
Складні реакції. Реакції, які відбуваються у декілька елементарних стадій, називаються складними. До них належать паралельні, консекутивні, спряжені, ланцюгові, послідовно-паралельні та ін.
Шляхии складних хімічних реакцій, в більшості випадків, відбуваються через прості моно- або бімолекулярні реакції.
Паралельними реакціями називаються реакції, в яких вихідна речовина бере участь одночасно в різних реакціях:
|
k1 A─ k2
|

 B
B
 D
DПрикладом таких реакцій є реакції утворення ізомерів фенолу, бензену при нітруванні нітратною(V) кислотою.
В живих організмах і рослинах глюкоза окиснюється до піровіноградної кислоти, а потім окиснення відбувається двома паралельними шляхами – по циклу Кребса і по циклу гексозомонофосфату.
При нагріванні оксиду нітрогену(ІІ) його розкладання відбувається за двома паралельними реакціями:
|
|
|
N2+O2 |
|
2NO |
|
|
|
|
N2O+O |




Для двох паралельних необоротних реакцій першого порядку можна записати:
![]()
Кожну з констант швидкості можна визначити, скориставшись рівняннями:
|
|
і |
|
Прямі
залежності
![]() і
і
![]() проходять через початок координат, з
кутовими коефіцієнтами, що дорівнюють
відповідно:
проходять через початок координат, з
кутовими коефіцієнтами, що дорівнюють
відповідно:
|
|
і |
|
Консекутивними або послідовними реакціями називаються хімічні реакції, в яких утворення кінцевого продукту з вихідних речовин відбувається через декілька проміжних стадій:
![]()
Проміжний продукт В іноді називається інтермедіатом.
Кінетика
послідовних реакцій залежить від
співвідношення значень констант
швидкості реакцій. На рис.3.11 приведені
залежності
![]() для послідовності двох реакцій першого
порядку при
для послідовності двох реакцій першого
порядку при
![]() і
і
![]() .
Значення
.
Значення
![]() і
і
![]() дістають із залежності
дістають із залежності
![]() і
і
![]() від
від
![]() .
.
|
|
|
Рис.3.11. Зміна концентрацій речовин А, В і D залежно
від
часу: а -
|

При
незначній відмінності між реакційною
здатністю реагентів А
і В ,
тобто коли
![]() ,
характер швидкості зміни концентрацій
речовин А, В
і D
в ході реакцій приведено на рис.3.12.
,
характер швидкості зміни концентрацій
речовин А, В
і D
в ході реакцій приведено на рис.3.12.
Як
бачимо, початкова швидкість зміни
концентрацій для речовин А
і
В максимальна.
Зменшення
![]() характеризує кут нахилу прямої
характеризує кут нахилу прямої
![]() .
Кути нахилу кривої залежності
.
Кути нахилу кривої залежності
![]() від
від
![]() на початковій і кінцевих стадіях
відповідають відповідно
на початковій і кінцевих стадіях
відповідають відповідно
![]() і
і
![]() .
Час, при якому
.
Час, при якому
![]() стає максимальним, визначають за
рівнянням:
стає максимальним, визначають за
рівнянням:
 (3.49)
(3.49)
|
|
|
Рис.3.12.
Залежність
реакцій
першого порядку при
|

На
початку реакції концентрація
![]() ,
а на кінцевій стадії кут нахилу кривої
,
а на кінцевій стадії кут нахилу кривої
![]() від
від
![]() дорівнює
дорівнює
![]() .
.
За механізмом послідовних реакцій відбуваються такі біологічні процеси, як гідроліз глікогену, гідроліз АТФ та ін.
Спряжені реакції – це хімічні реакції, в яких одна реакція відбувається тільки в присутності іншої. У біохімії такі реакції називають тандемними.
Прикладом спряженої реакції є реакції окиснення сульфату феруму(ІІ) пероксидом гідрогену:
![]() (І)
(І)
Одночасно з окисненням сульфату феруму(ІІ) відбувається і окиснення йодиду гідрогену:
![]() (ІІ)
(ІІ)
Самостійно окиснення йодиду гідрогену пероксидом гідрогену не відбувається, але в присутності сульфату феруму(ІІ), що окиснюється, окиснюється і йодид гідрогену.
В цієї спряженої реакції сульфат феруму(ІІ) є індуктором тому, що він індукує реакцію ІІ, а йодид гідрогену – акцептор. Сполуку пероксид гідрогену, яка є загальною для обох реакцій, називають актором.
Схематично спряжені реакції позначають:
![]() (І)
(І)
![]() (ІІ)
(ІІ)
де А – актор, В – індуктор, С – акцептор.
Ланцюгові реакції. Ланцюговий характер мають хімічні реакції, які перебігають за участю вільних атомів, радикалів, молекул, хімічно активних частинок, що мають надлишок енергії.
Приклади ланцюгової реакції є реакція утворення хлориду гідрогену з Гідрогену і Хлору:
1.
![]() Ініціювання, де hv
- квант світла.
Ініціювання, де hv
- квант світла.
2.
![]()
3.
![]() (v2
і
v3)
Зростання ланцюга.
(v2
і
v3)
Зростання ланцюга.
4.![]() (v4)
Перенос
ланцюга.
(v4)
Перенос
ланцюга.
5.
![]() (v5)
Обрив
ланцюга.
(v5)
Обрив
ланцюга.
За ланцюговим механізмом відбувається галогенування алканів:
![]()
![]()
![]() і
т.д.,
і
т.д.,
радіоактивне перетворення різних ізотопів:
|
|
тощо. |
Послідовно-паралельні хімічні реакції – це реакції, в яких одна з вихідних речовин бере участь як в реакціях утворення, так і в реакціях витрачання проміжних речовин. Типова схема таких реакцій має вигляд:
![]()
![]()
.............................
![]()
Кінетика ферментативних реакцій. Каталітична дія ферментів поділяється на три стадії: приєднання молекули субстрату (S) до ферменту (F); перетворення субстрату; відділення кінцевих продуктів реакції (R) від ферменту:
![]() (3.50)
(3.50)
де k1, k2, k3 i k4 – константи швидкості двох прямих і двох зворотніх реакцій.
За числом учасників ферментативних реакцій їх поділяють на одно- і двосубстратні. Найбільш поширені двосубстратні реакції з утворенням двох продуктів:
A + B L R1 + R2 .
Односубстратні реакції є мономолекулярними і відносяться до реакцій першого порядку. Більшість бімолекулярних реакцій є реакціями другого порядку. Їх швидкість пропорційна концентрації двох реагуючих речовин:
![]()
Швидкість ферментативної реакції залежить від активності ферменту. За одиницю будь-якого ферменту розуміють таку кількість ферменту, яка за певних умов ([S], pH, t) каталізує перетворення субстрату із швидкістю 1 моль/с. Ця одиниця називається катал- кат (1 кат = 109 нкат).
Активність ферменту виражають також у стандартних одиницях (кількість ферменту, яка каталізує перетворення 1 мкМ субстрату за 1 хвилину) (1 кат = 6 107 стандартних одиниць).
Залежність швидкості реакції від концентрації ферменту, як правило, є лінійною (рис.3.13 а, пряма 1) і порушується при нестачі субстрату чи активатору (крива 2).
Гіперболічний характер залежності V від [S] (рис.3.13 б) свідчить про те, що порядок ферментативної реакції змінюється: на початку це реакція першого порядку, в кінці – нульового, а при проміжних концентраціях субстрату – мішаного порядку.
|
а) |
б) |
|
Рис.3.13. Залежність початкової швидкості (V) реакції від концентрації ферменту (а) і субстрату (б) |
|


При вивченні кінетики ферментативних процесів Л. Міхаеліс і М. Ментен (пізніше Дж. Бриггс і Дж. Холдейн) розробили теорію кінетики дії ферментів для односубстратної реакції, яку виразили через рівняння:
![]() (3.51)
(3.51)
де Vmax – максимальна швидкість за умов насичення ферменту субстратом; Km – константа Міхаеліса, яка дорівнює:
![]()
Константа
Міхаеліса
![]() чисельно дорівнює концентрації субстрату,
при якій швидкість ферментативної
реакції досягає половини свого
максимального значення
Vmax
(рис.3.13, б).
чисельно дорівнює концентрації субстрату,
при якій швидкість ферментативної
реакції досягає половини свого
максимального значення
Vmax
(рис.3.13, б).
Рівняння (3.51) дійсно описує кінетичні данні рисунку (3.13, б). Так, при низьких концентраціях субстрату тобто, коли
![]()
швидкість реакції прямо пропорційна концентрації субстрату і описується рівнянням першого порядку
![]()
При високих концентраціях субстрату, коли
![]()
швидкість реакції максимальна і не залежить від концентрації субстрату
V =Vmax (реакція нульового порядку).
Якщо рівняння Міхаеліса-Ментена представити у вигляді подвійних обернених величин одержимо:
![]() (3.52)
(3.52)
Графічна
залежність
![]() від
від
![]() є прямою, яка відсікає на осі ординат
відрізок
є прямою, яка відсікає на осі ординат
відрізок
![]() ,
а на осі абсцис -
,
а на осі абсцис -
![]() (рис.3.14, а).
(рис.3.14, а).
|
|
|
|
|
а) б) в) |
||
|
Рис.3.14. Залежність оберненої швидкості від оберненої концентрації субстрату (а) [S]/V від [S] (б) і V від V/[S] (в) для визначення km i Vmax |
||


