Кишкун А.А
.pdf642 ■ Глава 9
всреднем до 12 нмоль/л (3−30 нмоль/л). В II триместре содержание 17ГПГ достигает 25 нмоль/л (20−35 нмоль/л), в III триместре — 35 нмоль/л (в амниотической жидкости — до 50 нмоль/л).
При подозрении на врождённый АГС исследуют концентрацию 17-ГПГ
вкрови из пуповины (или полученную в первые 3 дня после рождения). Референтные величины 17-ГПГ в крови новорождённых приведены в табл. 9-60.
Таблица 9-60. Референтные величины концентрации 17-ГПГ в крови новорождённых
Возраст |
Референтные величины |
||
|
|
||
нг/мл |
нмоль/л |
||
|
|||
|
|
|
|
Кровь из пуповины |
9−50 |
27,3−151,5 |
|
Недоношенные |
0,26−5,68 |
0,8−17,0 |
|
Новорождённые первые 3 дня |
0,07−0,77 |
0,2−2,3 |
|
|
|
|
|
Врождённая гиперплазия надпочечников, обусловленная недостаточностью 21-гидроксилазы, сопровождается повышением концентрации 17-ГПГ в крови до 40−220 нг/мл, при дефиците 11β-гидроксилазы повышение менее выражено. Более подробно о диагностике АГС см. в разделе «Функциональное состояние гипоталамо-гипофизарно-надпочечниковой системы».
ИММУНОРЕАКТИВНЫЙ ТРИПСИН В КРОВИ У НОВОРОЖДЁННЫХ (ТЕСТ НА ВРОЖДЁННЫЙ МУКОВИСЦИДОЗ)
Кистозный фиброз (муковисцидоз) — довольно распространённое заболевание. Муковисцидоз наследуется по аутосомно-рецессивному типу, его выявляют у 1 из 1500−2500 новорождённых. В связи с ранней диагностикой и эффективным лечением в настоящее время болезнь уже не считается присущей лишь детскому и юношескому возрасту. По мере совершенствования методов лечения и диагностики все большее количество больных достигают зрелого возраста. В настоящее время 50% больных доживают до 25 лет. Основной метод ранней постнатальной диагностики муковисцидоза — определение концентрации трипсина в сыворотке крови новорождённых. Референтные величины иммунореактивного трипсина (IRT) в сыворотке крови приведены в табл. 9-61 [Тиц Н., 1997].
Таблица 9-61. Референтные величины концентрации иммунореактивного трипсина в сыворотке крови
|
Возраст |
Иммунореактивный трипсин, мкг/л |
Кровь из пуповины |
23,3±1,9 |
|
0−6 |
мес |
31,3±5,4 |
6−12 мес |
37,1±6,9 |
|
1−3 |
года |
29,8±1,8 |
3−5 |
лет |
28,3±3,2 |
5−7 |
лет |
35,7±3,6 |
7−10 лет |
34,9±2,2 |
|
Взрослые |
33,3±11,1 |
|

Гормональные исследования ■ 643
Повышение концентрации трипсина в сыворотке крови детей в первые несколько недель после рождения свидетельствует о наличии муковисцидоза, в связи с чем определение этого показателя считают эффективным методом скрининга. По мере прогрессирования болезни и развития истинной недостаточности поджелудочной железы концентрация трипсина в сыворотке крови снижается.
ИССЛЕДОВАНИЕ КРОВИ НА ФЕНИЛКЕТОНЕМИЮ
Референтные величины концентрации фенилкетонов в крови у детей — до 121 мкмоль/л (до 2 мг%).
Нарушение метаболизма фенилаланина относится к весьма распространённым врождённым расстройствам метаболизма. Вследствие дефекта гена фенилаланин гидроксилазы (ФАГ-ген) развивается недостаточность фермента, и как следствие наступает блок в нормальном превращении фенилаланина в аминокислоту тирозин. Количество фенилаланина в организме накапливается, и концентрация его в крови увеличивается в 10−100 раз. Далее он превращается в фенилпировиноградную кислоту, оказывающую токсическое воздействие на нервную систему. В связи с этим большое значение имеет ранняя диагностика этого заболевания, так как длительное существование фенилкетонемии приводит к нарушению умственного развития ребёнка. Накопление фенилаланина в организме происходит постепенно и клиническая картина развивается медленно.
Исследование крови проводят в течение ближайших 48 ч (2−5 дней), после того как новорождённый получил молоко (источник фенилаланина). На чашку с питательной средой, засеянной бактериями фенилаланин-зависи- мого штамма Bacillus subtilis, накладывают диск из фильтровальной бумаги, смоченной несколькими каплями капиллярной крови, и контрольные диски, содержащие разное количество фенилаланина. Зона роста бактерий вокруг диска, смоченного кровью, пропорциональна концентрации фенилаланина в крови новорождённого.
Помимо дефекта фенилаланин гидроксилазы к гиперфенилаланинемии может приводить транзиторная тирозинемия новорождённых, которая, вероятно, возникает в результате неадекватного метаболизма тирозина. Основные типы гиперфенилаланинемии представлены в табл. 9-62.
Таблица 9-62. Типы гиперфенилаланинемии
|
Концентрация |
|
|
Тип |
фенилаланина |
Дефектный фермент |
Лечение |
|
в крови, мг% |
|
|
|
|
|
|
Классическая |
>20 |
Фенилаланин |
Диета |
фенилкетонурия |
|
гидроксилаза |
|
Атипичная |
12−20 |
Фенилаланин |
Диета |
фенилкетонурия |
|
гидроксилаза |
|
Персистирующая лёгкая |
2−12 |
Фенилаланин |
Диета |
гиперфенилаланинемия |
|
гидроксилаза |
|
Транзиторная гиперфенил- |
2−20 |
Неизвестен |
Нет |
аланинемия |
|
|
|

644 ■ Глава 9
Окончание табл. 9-62
Транзиторная тирозинемия |
2−12 |
Недостаточность |
Витамин С, |
|
|
β-гидроксифенил- |
смеси с |
|
|
пируват диоксиге- |
низким |
|
|
назы (и др.) |
содержани- |
|
|
Вторичная в ре- |
ем белка |
|
|
зультате недостатка |
|
|
|
витамина С |
|
Недостаточность дигидро- |
12−20 |
Дигидроптери- |
Допа, |
птеридин редуктазы |
|
дин редуктаза |
гидрокси- |
Дефекты синтеза биопте- |
|
|
триптофан |
12−20 |
Дигидроптеридин- |
Допа, |
|
рина |
|
синтетаза |
гидрокси- |
|
|
|
триптофан |
Основа лечения пациентов с недостаточностью фенилаланин гидроксилазы — ограничения фенилаланина в диете. При адекватно подобранной диете концентрация фенилаланина в крови не должна превышать верхний нормальный уровень или быть слегка ниже нормы.
У пациентов с транзиторной тирозинемией диета подбирается таким образом, чтобы концентрация тирозина в крови находилась в пределах между 0,5 и 1 мг% [Prieto J. et al., 1992].
ИССЛЕДОВАНИЕ КРОВИ НА ГАЛАКТОЗЕМИЮ
Референтные величины концентрации галактозы в крови у новорождённых — 0−1,11 ммоль/л (0−20 мг%), в более старшем возрасте — ниже 0,28 ммоль/л (5 мг%).
Воснове галактоземии лежит недостаточность галактозо-1-фосфат уридилтрансферазы (классическая галактоземия) или, реже, галактокиназы или галактозоэпимеразы.
Для скрининга на галактоземию применяют метод ингибирования роста бактерий. Исследование проводят у новорождённых, для чего берут кровь из пуповины или из пальца на фильтровальную бумагу. Ингибирование роста бактерий прямо пропорционально концентрации галактозы в крови (в норме ингибирования роста не происходит).
При количественном определении исследуют сыворотку крови или мочу. При наличии заболевания концентрация галактозы в крови может повышаться до 11,25 ммоль/л (300 мг%). Количественное определение галактозы в крови имеет важное значение для оценки адекватности подбора диеты больному. При правильно подобранной диете уровень галактозы в крови не должен превышать 0,15 ммоль/л (4 мг%). У здоровых новорождённых концентрация галактозы в моче ниже 3,33 ммоль/сут (60 мг/сут), впослед-
ствии — ниже 0,08 ммоль/сут (14 мг/сут). У больных галактоземией содержание галактозы в моче составляет 18,75−75 ммоль/л (500−2000 мг%).
Внастоящее время имеются диагностические наборы, позволяющие количественно определять активность галактозо-1-фосфат уридилтрансферазы в эритроцитах. Это исследование не только позволяет установить наличие недостаточности фермента, но и выявить гетерозиготных носителей дефектного гена.
Концентрация галактозы в крови также может повышаться при заболеваниях печени, гипертиреозе, нарушении пищеварения.

Глава 10
Генетические исследования
В последние годы прослеживается увеличение доли наследственных болезней в общей структуре заболеваний. В связи с этим возрастает роль генетических исследований в практической медицине. Без знания медицинской генетики невозможно эффективно проводить диагностику, лечение
ипрофилактику наследственных и врождённых заболеваний. Наследственная предрасположенность, вероятно, присуща практически
всем заболеваниям, но степень её значительно варьирует. Если рассматривать роль наследственных факторов в возникновении различных заболеваний, то можно выделить следующие их группы.
■Заболевания, происхождение которых полностью определяется генетическими факторами (воздействием патологического гена); к этой группе относятся моногенные болезни, наследование которых подчиняется основным правилам законов Менделя (менделирующие заболевания), а воздействие внешней среды может оказывать влияние лишь на интенсивность тех или иных проявлений патологического процесса (на его симптоматику).
■Болезни, возникновение которых определяется в основном воздействием внешней среды (инфекции, травмы и т.п.); наследственность может лишь влиять на некоторые количественные характеристики реакции организма, определять особенности течения патологического процесса.
■Заболевания, при которых наследственность является причинным фактором, но для его проявления необходимы определённые воздействия внешней среды, их наследование не подчиняется законам Менделя (неменделирующие заболевания); они получили название мультифакториальных.
Наследственные заболевания
Развитие каждого индивида — результат взаимодействия генетических и внешнесредовых факторов. Набор генов человека устанавливается при оплодотворении и затем вместе с факторами внешней среды определяет особенности развития. Совокупность генов организма получила название генома. Геном в целом весьма стабилен, но под влиянием меняющихся условий внешней среды в нём могут происходить изменения — мутации.
Основные единицы наследственности — гены (участки молекулы ДНК). Механизм передачи наследственной информации основан на способности ДНК к самоудвоению (репликации). ДНК содержит генетический код (система записи информации о расположении аминокислот в белках с помощью последовательности расположения нуклеотидов в ДНК и информационной РНК), который определяет развитие и метаболизм клеток. Гены расположены в хромосомах, структурных элементах ядра клетки, содержащих ДНК. Место, занимаемое геном, называется локусом. Моногенные

646 ■ Глава 10
заболевания — монолокусные, полигенные болезни (мультифакториальные) — мультилокусные.
Хромосомы (видимые в световом микроскопе палочковидные структуры в ядрах клеток) состоят из многих тысяч генов. У человека каждая соматическая, то есть не половая, клетка содержит 46 хромосом, представленных 23 парами. Одна из пар — половые хромосомы (Х и Y) — определяет пол индивида. В ядрах соматических клеток у женщин присутствуют две хромосомы X, у мужчин — одна хромосома Х и одна хромосома Y. Половые хромосомы мужчин гетерологичны: хромосома X больше, в ней содержится множество генов, отвечающих как за определение пола, так и за другие признаки организма; хромосома Y маленькая, имеет отличающуюся от хромосомы X форму и несёт главным образом гены, детерминирующие мужской пол. Клетки содержат 22 пары аутосом. Аутосомные хромосомы человека разделяют на 7 групп: А (1-, 2-, 3-я пары хромосом), В (4-, 5-я пары), С (6-, 7-, 8-, 9-, 10-, 11-, 12-я пары, а также хромосома Х, по размерам сходная с хромосомами 6 и 7), D (13-, 14-, 15-я пары), Е (16-, 17-, 18-я пары), F (19-, 20-я пары), G (21-, 22-я пары и хромосома Y).
Гены расположены вдоль хромосом линейно, причём каждый ген занимает строго определённое место (локус). Гены, занимающие гомологичные локусы, называются аллельными. Каждый человек имеет по два аллеля одного и того же гена: по одному на каждой хромосоме каждой пары, за исключением большинства генов на хромосомах X и Y у мужчин. В тех случаях, когда в гомологичных участках хромосомы присутствуют одинаковые аллели, говорят о гомозиготности, когда же в них содержатся разные аллели одного и того же гена, то принято говорить о гетерозиготности по данному гену. Если ген (аллель) проявляет свой эффект, присутствуя только в одной хромосоме, он называется доминантным. Рецессивный ген проявляется только в том случае, если он присутствует у обоих членов хромосомной пары (или в единственной хромосоме X у мужчин или у женщин с генотипом Х0). Ген (и соответствующий ему признак) называется Х-сцепленным, если он локализуется в хромосоме X. Все остальные гены называются аутосомными.
Различают доминантный и рецессивный тип наследования. При доминантном наследовании признак проявляется как в гомозиготном, так и в гетерозиготном состояниях. При рецессивном наследовании фенотипические (совокупность внешних и внутренних признаков организма) проявления наблюдают только в гомозиготном состоянии, в то время как при гетерозиготности они отсутствуют. Возможен также сцепленный с полом доминантный или рецессивный тип наследования; таким образом наследуются признаки, связанные с генами, локализованными в половых хромосомах.
При доминантно-наследуемых заболеваниях обычно поражаются несколько поколений одной семьи. При рецессивном наследовании в семье может длительно существовать скрытое гетерозиготное носительство мутантного гена, в связи с чем больные дети могут рождаться у здоровых родителей или даже в семьях, в которых в нескольких поколениях данное заболевание отсутствовало.
В основе наследственных болезней лежат мутации генов. Понимание мутаций невозможно без современного понимания термина «геном». В настоящее время геном рассматривается как мультигеномная симбиотическая

Генетические исследования ■ 647
конструкция, состоящая из облигатных и факультативных элементов. Основу облигатных элементов составляют структурные локусы (гены), количество и расположение которых в геноме достаточно постоянны. На долю структурных генов приходится приблизительно 10−15% генома. Понятие «ген» включает транскрибируемую область: экзоны (собственно кодирующий участок) и интроны (некодирующий, разделяющий экзоны участок);
ифланкирующие последовательности — лидерная, предшествующая нача-
лу гена, и хвостовая нетранслируемая область. Факультативные элементы (85−90% от всего генома) представляют собой ДНК, которая не несёт информации об аминокислотной последовательности белков и не является строго обязательной. Эта ДНК может участвовать в регуляции экспрессии генов, выполнять структурные функции, повышать точность гомологичного спаривания и рекомбинации, способствовать успешной репликации ДНК. Участие факультативных элементов в наследственной передаче признаков и формировании мутационной изменчивости в настоящее время доказано. Такое сложное строение генома определяет разнообразие генных мутаций.
Всамом широком смысле мутация — устойчивое, передаваемое по наследству изменение в ДНК. Мутации могут сопровождаться видимыми при микроскопии изменениями структуры хромосом: делеция — выпадение участка хромосомы; дупликация — удвоение участка хромосомы, инсерция (инверсия) — разрыв участка хромосомы, поворот его на 180° и прикрепление к месту разрыва; транслокация — отрыв участка одной хромосомы
иприкрепление его к другой. Такие мутации обладают наибольшим повреждающим действием. В других случаях мутации могут заключаться в замене одного из пуриновых или пиримидиновых нуклеотидов единичного гена (точечные мутации). К таким мутациям относятся: миссенс-мутации (мутации с изменением смысла) — замена нуклеотидов в кодонах с фенотипическими проявлениями; нонсенс-мутации (бессмысленные) — замены нуклеотидов, при которых образуются терминирующие кодоны, в результате синтез кодируемого геном белка преждевременно обрывается; сплайсинговые мутации — замены нуклеотидов на стыке экзонов и интронов, что приводит к синтезу удлинённых молекул белка.
Относительно недавно выявлен новый класс мутаций — динамические мутации или мутации экспансии, связанные с нестабильностью количества тринуклеотидных повторов в функционально значимых частях генов. Многие тринуклеотидные повторы, локализованные в транскрибируемых или регуляторных областях генов, характеризуются высоким уровнем популяционной изменчивости, в пределах которого не наблюдается фенотипических нарушений (то есть болезнь не развивается). Болезнь развивается лишь тогда, когда количество повторов в этих сайтах превосходит определённый критический уровень. Такие мутации не наследуются в соответствии с законом Менделя.
Таким образом, наследственные болезни — это болезни, обусловленные повреждениями генома клетки, которые могут затрагивать весь геном, отдельные хромосомы и вызывать хромосомные болезни, или затрагивать отдельные гены и быть причиной генных болезней.
Все наследственные болезни принято разделять на три большие группы [Беркоу Р., 1997]:
■ моногенные;

648 ■ Глава 10
■полигенные, или мультифакториальные, при которых мутации нескольких генов и негенетические факторы взаимодействуют;
■хромосомные нарушения, или аномалии в структуре или количестве хромосом.
Заболевания, относящиеся к двум первым группам, часто называют генетическими, а к третьей — хромосомными болезнями (табл. 10-1).
Таблица 10-1. Классификация наследственных заболеваний
Хромосомные |
Моногенные |
Мультифакториальные |
|
(полигенные) |
|||
|
|
||
|
|
|
|
Аномалии количества |
Аутосомно-доминантные: |
ЦНС: некоторые |
|
Половых хромосом: |
синдром Марфана; |
формы эпилепсии, |
|
− синдром Шерешев- |
болезнь фон Вил- |
шизофрения и др. |
|
Сердечно-сосудистой |
|||
ского−Тернера; |
лебранда; |
||
− синдром Кляйнфел- |
анемия Минковско- |
системы: ревматизм, |
|
гипертоническая |
|||
тера; |
го−Шоффара и др |
||
болезнь, атеросклероз |
|||
− синдром трисомии Х; |
Аутосомно-рецессивные: |
||
и др. |
|||
− синдром 47, ХYY |
− фенилкетонурия; |
Кожи: атопический |
|
Аутосом: |
− галактоземия; |
дерматит, псориаз и др. |
|
− болезнь Дауна; |
− муковисцидоз и др. |
Дыхательной системы: |
|
− синдром Эдвардса; |
Х-сцепленные рецессив- |
бронхиальная астма, |
|
− синдром Патау; |
ные: |
аллергический альве- |
|
− частичная трисомия |
гемофилия А и B; |
олит и др. |
|
Мочевой системы: |
|||
22 |
миопатия Дюшена; |
||
мочекаменная болезнь, |
|||
Структурные аномалии хро- |
и др. |
||
энурез и др. |
|||
мосом: |
Х-сцепленные доминант- |
||
Пищеварительной |
|||
синдром «кошачьего |
ные: |
||
системы: язвенная |
|||
крика»; |
− витамин D-резис- |
||
болезнь, неспецифи- |
|||
синдром 4p-делеции; |
тентный рахит; |
||
ческий язвенный колит |
|||
синдромы микроделе- |
− коричневая окраска |
и др. |
|
ций соседних генов |
эмали зубов и др. |
|
Хромосомные болезни могут быть обусловлены количественными аномалиями хромосом (геномные мутации), а также структурными аномалиями хромосом (хромосомные аберрации). Клинически почти все хромосомные болезни проявляются нарушением интелектуального развития и множественными врождёнными пороками, часто несовместимыми с жизнью.
Моногенные болезни развиваются вследствие повреждения отдельных генов. К моногенным болезням относятся большинство наследственных болезней обмена (фенилкетонурия, галактоземия, мукополисахаридозы, муковисцидоз, АГС, гликогенозы и др.). Моногенные болезни наследуются в соответствии с законами Менделя и по типу наследования могут быть разделены на аутосомно-доминантные, аутосомно-рецессивные и сцепленные с хромосомой X.
Мультифакториальные болезни являются полигенными, для их развития необходимо влияние определённых факторов внешней среды. Общие признаки мультифакториальных заболеваний следующие.
■ Высокая частота среди населения.

Генетические исследования ■ 649
■Выраженный клинический полиморфизм.
■Сходство клинических проявлений у пробанда и ближайших родственников.
■Возрастные и половые различия.
■Более раннее начало и некоторое усиление клинических проявлений в нисходящих поколениях.
■Вариабельная терапевтическая эффективность ЛС.
■Сходство клинических и других проявлений болезни у ближайших
родственников и пробанда (коэффициент наследуемости для мультифакториальных заболеваний превышает 50−60%).
■Несоответствие закономерностей наследования законам Менделя. Для клинической практики важно понимать сущность термина «врож-
дённые пороки развития», которые могут быть одиночными или множественными, наследственными или спорадическими. К наследственным болезням нельзя относить те врождённые заболевания, которые возникают в критические периоды эмбриогенеза под воздействием неблагоприятных факторов внешней среды (физических, химических, биологических и др.) и не передаются по наследству. Примером такой патологии могут быть врождённые пороки сердца, которые часто обусловлены патологическими воздействиями в период закладки сердца (I триместр беременности), например вирусной инфекцией, тропной к тканям формирующегося сердца; алкогольный синдром плода, аномалии развития конечностей, ушных раковин, почек, пищеварительного тракта и др. В таких случаях генетические факторы формируют только наследственную предрасположенность или повышенную восприимчивость к действию определённых факторов внешней среды. По данным ВОЗ аномалии развития присутствуют у 2,5% всех новорождённых; 1,5% из них обусловлены действием неблагоприятных экзогенных факторов во время беременности, остальные имеют преимущественно генетическую природу. Разграничение наследственных и врождённых заболеваний, которые не передаются по наследству, имеет очень важное практическое значение для прогнозирования потомства в данной семье.
Методы диагностики наследственных заболеваний
В настоящее время практическая медицина имеет целый арсенал диагностических методов, позволяющих с определённой вероятностью выявлять наследственные заболевания. Диагностическая чувствительность и специфичность этих методов различна — одни позволяют только предположить наличие заболевания, другие с большой точностью выявляют мутации, лежащие в основе болезни или определяющие особенности её течения.
Цитогенетические методы
Цитогенетические методы исследования применяют для диагностики хромосомных болезней. Они включают:
■исследования полового хроматина — определение Х- и Y-хроматина;
■кариотипирование (кариотип — совокупность хромосом клетки) — определение количества и структуры хромосом с целью диагностики хромосомных болезней (геномных мутаций и хромосомных аберраций).

650 ■ Глава 10
ОПРЕДЕЛЕНИЕ Х- И Y-ХРОМАТИНА
Определение Х- и Y-хроматина часто называют методом экспресс-диа- гностики пола. Исследуют клетки слизистой оболочки ротовой полости, вагинального эпителия или волосяной луковицы. В ядрах клеток женщин в диплоидном наборе присутствуют две хромосомы Х, одна из которых полностью инактивирована (спирализована, плотно упакована) уже на ранних этапах эмбрионального развития и видна в виде глыбки гетерохроматина, прикреплённого к оболочке ядра. Инактивированная хромосома Х называется половым хроматином или тельцем Барра. Для выявления полового Х-хро- матина (тельца Барра) в ядрах клеток мазки окрашивают ацетарсеином и препараты просматривают с помощью обычного светового микроскопа. В норме у женщин обнаруживают одну глыбку Х-хроматина, а у мужчин её нет.
Для выявления мужского Y-полового хроматина (F-тельце) мазки окрашивают акрихином и просматривают с помощью люминисцентного микроскопа. Y-хроматин выявляют в виде сильно светящейся точки, по величине и интенсивности свечения отличающейся от остальных хромоцентров. Он обнаруживается в ядрах клеток мужского организма.
Отсутствие тельца Барра у женщин свидетельствует о хромосомном заболевании — синдроме Шерешевского−Тернера (кариотип 45, Х0). Присутствие у мужчин тельца Барра свидетельствует о синдроме Кляйнфелтера (кариотип 47, ХХY).
Определение Х- и Y-хроматина — скрининговый метод, окончательный диагноз хромосомной болезни ставят только после исследования кариотипа.
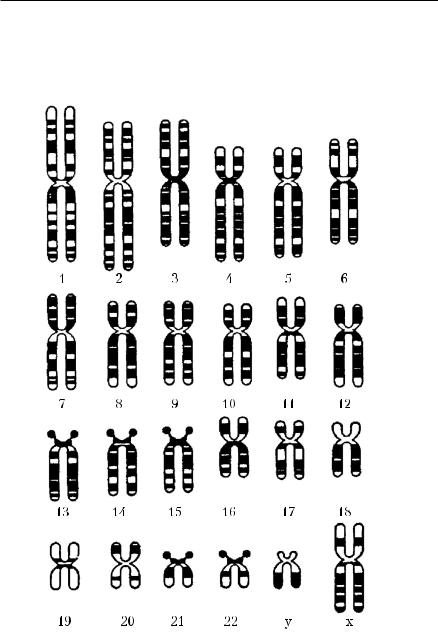
КАРИОТИПИРОВАНИЕ
Для изучения хромосом чаще всего используют препараты кратковременной культуры крови, а также клетки костного мозга и культуры фибробластов. Доставленную в лабораторию кровь с антикоагулянтом подвергают центрифугированию для осаждения эритроцитов, а лейкоциты инкубируют
вкультуральной среде 2−3 дня. К образцу крови добавляют фитогемагглютинин, так как он ускоряет агглютинацию эритроцитов и стимулирует деление лимфоцитов. Наиболее подходящая фаза для исследования хромосом — метафаза митоза, поэтому для остановки деления лимфоцитов на этой стадии используют колхицин. Добавление этого препарата к культуре приводит к увеличению доли клеток, находящихся в метафазе, то есть
втой стадии клеточного цикла, когда хромосомы видны лучше всего. Каждая хромосома реплицируется (производит свою копию) и после соответствующей окраски видна в виде двух хроматид, прикреплённых к центромере, или центральной перетяжке. Затем клетки обрабатывают гипото-
ническим раствором хлорида натрия, фиксируют и окрашивают.
Для окраски хромосом чаще используют краситель Романовского−Гимзы, 2% ацеткармин или 2% ацетарсеин. Они окрашивают хромосомы целиком, равномерно (рутинный метод) и могут быть использованы для выявления численных аномалий хромосом человека.
Для получения детальной картины структуры хромосом, идентификации (определения) отдельных хромосом или их сегментов используют различные способы дифференциального окрашивания. Наиболее часто применяют методы Гимза, а также G- и Q-бендинга. При микроскопии препа-