

6.4. Люмінесцентний аналіз.
Люмінесцентний аналіз базується на вимірюванні інтенсивності люмінесценції речовини, яку треба визначити. Люмінесценцією називають «холодне» світіння, тобто таке, яке не пов’язане з нагріванням речовини. Молекула випромінює енергію, яку вона отримала, збуджуючись від катодних променів (катодолюмінесценція), від хімічної реакції (хемілюмінесценція), від біохімічних процесів у живому організмі (біолюмінесценція) та ін. Найбільше в аналізі використовують фотолюмінесценцію, коли збудження відбувається завдяки поглинанню квантів світла з більш високою енергією, ніж потім випромінюється. Фото-люмінесценція, яка продовжується деякий час після припинення збудження, називається фосфоресценцією. У розчинах можлива тільки флуоресценція, що зникає, як тільки припиняється збудження. Метод кількісного аналізу, що базується на вимірюванні інтенсивності флуоресценції залежно від концентрації речовини у розчині, називають флуориметрією. Цей метод має високу чутливість, тому його використовують для визначення речовин, які містяться в аналізованій пробі у дуже малих кількостях, але мають велике значення, приміром, вітамінів. Шкідливі речовини, які проявляють здатність до люмінесценції, також визначають цим методом.
6.4.1. Явище люмінесценції. Спрощено явище фотолюмінесценції можна описати так:
МОЛЕКУЛА + СВІТЛО ----------------- МОЛЕКУЛА
(Основний стан) (Висока енергія) (Поглинання→збудження) (Збуджений стан)
МОЛЕКУЛА -------------------- МОЛЕКУЛА + СВІТЛО
(Збуджений стан) Випромінювання (Основний стан) (Більш низька енергія)

Рис.6.4.1. Електронні переходи у молекулі
П оглинання
ультрафіолетового або видимого світла
викликає електронний перехід (10-15с),
внаслідок якого молекула збуджується:
переходить з основного електронного
стану на один з підрівнів першого
збудженого електронного стану (рис.6.4.1).
Далі без випромінювання переходить
(10-12с)
на найнижчий підрівень першого збудженого
стану, а потім (10-8с)
повертається на один з підрівнів
основного стану – при цьому виникає
флуоресцентне випромінювання. Отже,
частина енергії, що поглинається на
етапі збудження, втрачається без
випромінювання (переходить у теплові
коливання). Це пояснює правило
Стокса
(1852): довжина
хвилі люмінесценції більша за довжину
хвилі збуджувального світла
(що поглинається). Нагадаємо, що більша
довжина хвилі відповідає меншій енергії
квантів, що випромінюються.
оглинання
ультрафіолетового або видимого світла
викликає електронний перехід (10-15с),
внаслідок якого молекула збуджується:
переходить з основного електронного
стану на один з підрівнів першого
збудженого електронного стану (рис.6.4.1).
Далі без випромінювання переходить
(10-12с)
на найнижчий підрівень першого збудженого
стану, а потім (10-8с)
повертається на один з підрівнів
основного стану – при цьому виникає
флуоресцентне випромінювання. Отже,
частина енергії, що поглинається на
етапі збудження, втрачається без
випромінювання (переходить у теплові
коливання). Це пояснює правило
Стокса
(1852): довжина
хвилі люмінесценції більша за довжину
хвилі збуджувального світла
(що поглинається). Нагадаємо, що більша
довжина хвилі відповідає меншій енергії
квантів, що випромінюються.
Кожна речовина, яку визначають флуориметрично, має свій спектр поглинання і випромінювання, тобто залежність інтенсивності (І) поглинання або випромінювання від довжини хвилі або частоти (рис.6.4.2).
Рис. 6.4.2. Правило Стокса
Спектри люмінесценції подібні до спектрів поглинання (збудження). Якщо по осі абсцис відкладати частоту, а не довжину хвилі, то для багатьох складних молекул спектри поглинання і флуоресценції є дзеркальним відбиттям один одного (правило Лєвшина), при цьому спектр флуоресценції, за правилом Стокса, зміщений у бік менших частот (більших довжин хвиль). Відстань між максимумами спектрів поглинання і флуоресценції називається стоксовим зміщенням. Це зміщення дає змогу відділити флуоресцентне випромінювання від збуджувального світла (за допомогою світлофільтрів) і зробити надійнішим визначення речовини люмінесцентним методом.
Збуджена молекула може перейти в основний стан і без випромінювання, зайва енергія при цьому перетворюється у теплові коливання. Відношення числа квантів флуоресценції (що випромінюється) до числа квантів, що поглинаються на етапі збудження, називається квантовим виходом фотолюмінесценції. За законом С.І.Вавілова “фотолюмінесценція може зберігати постійний квантовий вихід, якщо збуджувальна хвиля перетворюється у довшу, ніж вона сама”.
За умови постійного квантового виходу інтенсивність флуоресценції прямо пропорційна концентрації речовини, що світиться, тобто вона може бути аналітичним сигналом для визначення цієї речовини. Але така залежність спостерігається, якщо концентрація дуже мала (10-9–10-6 моль/л). Зі збільшенням концентрації речовини, що світиться, інтенсивність світіння спочатку збільшується пропорційно концентрації, потім починає “відставати” від концентрації і навіть зменшуватись за подальшого збільшення концентрації (рис.6.4.3). Це явище називається концентраційним гасінням флуоресценції. Воно має зворотний характер: при розведенні розчинів світіння відновлюється, що свідчить про відсутність складних фізико-хімічних перетворень. Для більшості речовин концентраційним бар’єром є 10-5–10-4 М. Це відповідає такій відстані між молекулами, яка значно менша довжини хвилі світла, що випромінюється, але більша за розміри молекул, що випромінюють. У таких умовах явище гасіння добре описується теорією міграції енергії, відповідно до якої випромінюване світло знову поглинається іншими такими самими молекулами. (Крім концентраційного існують ще температурне гасіння та гасіння люмінесценції сторонніми речовинами).
 Рис.6.4.3.
Концентраційне гасіння люмінесценції
Рис.6.4.3.
Концентраційне гасіння люмінесценції
Важливою перевагою флуориметрії (молекулярно-емісійної спектрофотометрії) над молекулярно-абсорбційною спектрофотометрією є більша чутливість методу (чутливість – це найменша кількість або концентрація речовини, яку можна визначити даним методом). У молекулярно-абсорбційній спектрофотометрії вимірюється зменшення інтенсивності на фоні інтенсивного світлового потоку тієї самої довжини хвилі. У флуориметрії вимірюється світіння, яке відділяється від збуджувального випромінювання, тобто фон можна зменшити практично до нуля. При цьому інтенсивність слабкого сигналу вимірюється точніше. Молекулярно-абсорбційна спектрофотометрія дає змогу визначати до 10-5 моль/л або 10-6 г/мл (1 мкг/мл). Флуориметрію використовують для визначення концентрацій до 10-8 моль/л або 10-9 г/мл (1 нг/мл). Але на відміну від молекулярної абсорбції флуориметрія не може визначати великі концентрації.
6.4.2. Об’єкти аналізу. Для визначення якості харчових продуктів, сортування насіння, виявлення фальсифікацій тощо застосовують “сортовий люмінесцентний аналіз”. Це візуальне спостереження власної люмінесценції обєктів і люмінесценції спеціальних барвників, якими обробляють досліджуваний обєкт.
Кількісні флуориметричні методи застосовують для визначення біологічно активних речовин: вітамінів, антибіотиків, гормонів та ін. За способом визначення ці речовини можна поділити на дві групи: вимірюють власне світіння речовини (рибофлавін), або речовину, яка не світиться (тіамін) переводять у речовину, яка має таку здатність (тіохром), виконуючи певну аналітичну реакцію.
До першої групи належать вітаміни А і В2. У неводних розчинах ретінол (вітамін А) проявляє здатність до флуоресценції з максимумом випромінювання при 480 нм, якщо збуджується ультрафіолетовими променями з довжиною хвилі 330 – 360 нм. Рибофлавін (вітамін В2) у нейтральних водних розчинах має власну жовто-зелену флуоресценцію: поглинає світло з довжиною хвилі 350 – 480 нм, випромінює – 510 – 650 нм.
До другої групи належать вітаміни В1 і С. Тіамін (вітамін В1) окиснюється у лужному середовищі у тіохром – жовту сполуку, яка екстрагується неводними розчинниками і проявляє інтенсивну синю флуоресценцію при опромінюванні ультрафіолетом: поглинає світло з довжиною хвилі 320 – 390 нм, випромінює – 400 – 580 нм. Вітамін С (аскорбінова кислота) в реакції з о-фенілендіаміном утворює хіноксалін, який дає максимальну флуоресценцію при 430 нм, якщо збуджується квантами з довжиною хвилі 350 нм.
Поліциклічні ароматичні вуглеводні, які мають канцерогенні властивості і є небезпечними забрудниками зовнішнього середовища і харчових продуктів, визначають завдяки їхній здатності до флуоресценції. Метод визначення 3,4-бензпірену базується на тому, що в розчині сульфатної кислоти він знаходиться у вигляді катіону, який поглинає світло при 520 нм і випромінює при 545 нм.
6.4.3. Умови визначення. Незначні коливання температури помітно не впливають на інтенсивність флуоресценції. При значному підвищенні температури може спостерігатися температурне гасіння. При глибокому охолоджуванні аж до температури рідкого азоту (-195оС) значно збільшується квантовий вихід люмінесценції, виникає люмінесценція речовин, які не світяться при звичайних температурах.
Важливою умовою правильності флуориметричного визначення є кислотність розчину. Більшість речовин, які визначають, є слабкими кислотами або основами, тому форма їхнього існування у розчині (і відповідно здатність до люмінесценції) дуже залежить від рН розчину. Чутливість і точність визначення значно погіршуються у присутності багатьох сторонніх речовин, тому треба використовувати посуд, реактиви і розчинники високої чистоти. Сторонні речовини можуть значно знижувати інтенсивність флуоресценції. Гасіння сторонніми речовинами спостерігається як під дією неорганічних іонів (І¯, Br¯,Cu2+, Fe3+), так і органічних сполук (анілін, гідрохінон та ін.). Причиною значних помилок може бути присутність розчиненого кисню – він гасить флуоресценцію багатьох ароматичних спролук. Азот, аргон та інші інертні гази не впливають на флуоресценцію. Насичуючи розчин азотом, можна видалити розчинений кисень.
6.4.4. Флуориметр. Вимірювання аналітичного сигналу. Будь-який флуориметричний прилад складається з таких основних частин: 1)джерело збуджувального випромінювання; 2)первинний світлофільтр (або монохроматор) для цього випромінювання; 3)кювета (пробірка), де міститься речовина, що світиться під дією збуджувального випромінювання; 4)вторинний світлофільтр (або монохроматор) для флуоресцентного випромінювання; 5)фотоелемент (або фотопомножувач), який перетворює світловий потік в електричний сигнал. Найпростішими є прилади для візуального якісного аналізу і люмінесцентного титрування – приймачем випромінювання в них є очі лаборанта-аналітика. Найскладнішими є прилади для вивчення спектрів та інших кількісних характеристик люмінесцентного випромінювання – так звані спектрофлуориметри. Джерелом збуджувального випромінювання найчастіше є ртутно-кварцева лампа. В ній випромінюють атоми ртуті, які дають багато інтенсивних ліній як у видимій, так і в ультрафіолетовій частині спектра. Колба лампи зроблена з кварцу, який краще пропускає ультрафіолетове випромінювання, ніж звичайне скло.
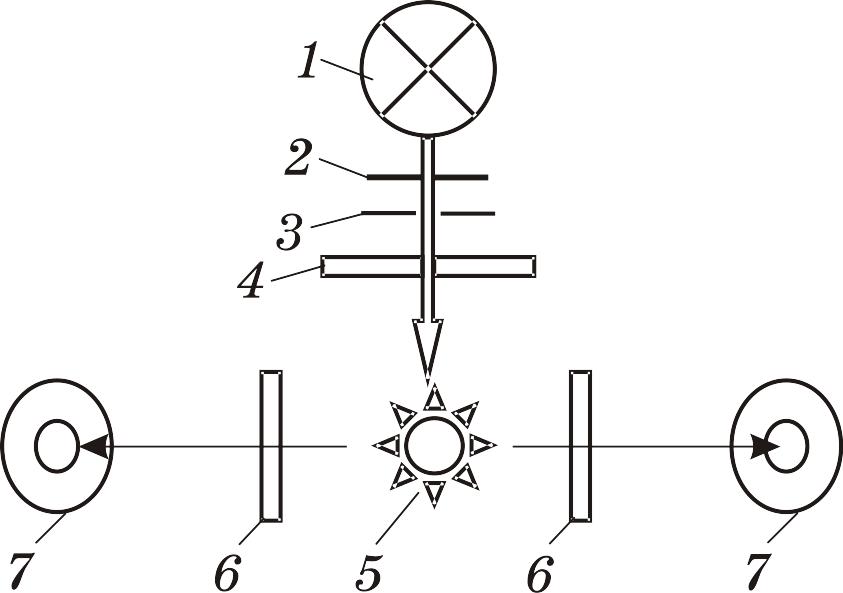 Рис
6.4.4. Принципова схема флуориметра:
Рис
6.4.4. Принципова схема флуориметра:
1 - ртутно-кварцева лампа; 2 – заслонка; 3 – діафрагма; 4 – первинний світлофільтр 5 – пробірка (кювета) з розчином, що світиться; 6 –вторинні світлофільтри; 7 – фотоелементи.
Найчастіше для флуориметричних визначень використовують електронний флуориметр типу ЭФ-3М (рис.6.4.4). Первинний і вторинний світлофільтри підбираються залежно від того, яку речовину визначають на флуориметрі. Максимум пропускання первинного світлофільтра повинен співпадати з максимумом поглинання цієї речовини, а максимум пропускання вторинного світлофільтра повинен співпадати з максимумом флуоресцентного випромінювання (див. рис. 6.4.1 – правило Стокса). Стоксове зміщення дає можливість відділити флуоресценцію від збуджувального випромінювання. З цією ж метою інтенсивність флуоресценції реєструють у напрямку, перпендикулярному до напрямку збуджувального випромінювання. Ці заходи дають можливість зменшити фон практично до нуля і збільшити точність вимірювання.
6.5. Інші оптичні методи аналізу. 6.5.1. Поляриметрія. Відповідно до електромагнітної теорії світла електромагнітні коливання у природному пучку світла здійснюється у всіх площинах, перпендикулярних до напрямку руху променя (поперечні коливання). Якщо поперечні коливання світлових хвиль здійснюється тільки в одній площині, то такий промінь світла називається поляризованим. Площина, в якій здійснюється коливання променя, називається площиною коливання поляризованого променя, а площина, перпендикулярна до неї, – площиною поляризації. При пропусканні променя поляризованого світла через оптично неактивну речовину ніяких змін у напрямку коливань не відбувається, і вони продовжують коливатись у тій же площині. Якщо промінь поляризованого світла пропустили через оптично активну речовину, то на виході з неї коливання в поляризованому промені будуть проходити вже в іншій площині, розташованій відносно первинної під деяким кутом. Цей кут називається кутом обертання площини поляризації. Він залежить від концентрації оптично активної речовини у досліджуваному розчині. До цієї категорії сполук належать органічні речовини, які містять у своїй молекулі асиметричні атоми карбону. Це глюкоза, винна кислота, морфін тощо. Метод аналізу, який ґрунтується на вимірюванні кута обертання площини поляризації променя світла, що пройшов через оптично активне середовище, називається поляриметричним.
6.5.2. Рефрактометрія. Промінь світла, що переходить з одного середовища до іншого, частково відбивається від поверхні розділу, а частково переходить до іншого середовища, змінюючи при цьому свій початковий напрямок. Зміну напрямку прямолінійного поширення світла при переході з одного середовища до іншого називають заломленням або рефракцією. Метод аналізу, що ґрунтується на визначенні показника заломлення досліджуваного розчину залежно від концентрації, називається рефрактометричним.
6.5.3. Нефелометрія і турбідіметрія. Нефело– і турбідиметричний методи аналізу базуються на тому, що компонент, який визначається, переводять у малорозчинну сполуку, яка перебуває у вигляді зависі і здатна





І0 Іп
Ір
Рис.6.5.1. Схема вимірюваня світлових потоків у нефелометрії (Ір) і у турбідиметрії (Іп).
розсіювати світло. Під нефелометричною (турбідиметричною) зависсю розуміють суспензії малорозчинних речовин у сильнорозведених розчинах, які розсіюють світло протягом часу, достатнього для вимірювання. При проходженні пучка світла І0 через дрібні тверді частинки, що містяться в розчині у вигляді зависі (суспензії), частина світлової енергії Ір розсіюється, а частина Іп проходить через кювету (рис. 6.5.1). На основі закону збереження енергії можна записати: І0 = Іп + Ір
Значення обох членів Іп і Ір у рівнянні залежить від концентрації завислих частинок у розчині. Якщо вміст речовини знаходять за інтенсивністю розсіяного світла Ір, то такий метод називають нефелометричним. Метод вивчення вмісту речовини за послабленістю світлового потоку Іп називається турбідиметрією.
7. Електрохімічні методи аналізу базуються на вимірюванні електричних параметрів електрохімічних процесів, що виникають у досліджуваному розчині залежно від його концентрації. Таке вимірювання здійснюється за допомогою електрохімічної комірки. Це посуд з досліджуваним розчином, у який занурені електроди, підключені до вимірювального приладу.
7.1.Потенціометрія – один з електрохімічних методів аналізу, який базується на вимірюванні потенціалу індикаторного електроду. Індикаторним називають електрод, потенціал якого лінійно залежить від логарифму концентрації саме тих іонів, які треба визначити у розчині, і не залежить від концентрації усіх інших іонів, присутніх у тому ж розчині. Цим методом можна визначити такі іони у досліджуваному розчині, для яких є індикаторний електрод. Для визначення рН розчину (pH=-lgH+) використовують скляний електрод, індикаторний до іонів Н+. Для визначення NO3- використовують нітрат-селективний індикаторний електрод.
Але виміряти потенціал окремо взятого індикаторного електроду неможливо. Щоб виміряти потенціал цього електроду у розчин занурюють ще один (допоміжний) електрод порівняння. Це електрод, потенціал якого відомий і не змінюється протягом вимірювань. Індикаторний і допоміжний електроди підключають до потенціометра і вимірюють ЕРС побудованого з них гальванічного елемента. У цьому випадку ЕРС, що вимірюється, як і потенціал індикаторного електроду, лінійно залежить від логарифму концентрації іонів, що визначаються. 7.1.1.Класифікація електродів за хімічною будовою.
Електроди називають індикаторним і електродом порівняння залежно від того, як вони використовуються у даному вимірюванні. Залежно від хімічної будови електроди поділяються на електроно-обмінні і іоно-обмінні.
7.1.1.1.Електронообмінними називають електроди, на поверхні яких відбувається певна реакція за участю електронів і саме вона визначає потенціал за рівнянням Нернста.
1)Електронообмінні електроди І роду – це металева дротинка (пластинка), занурена у розчин солі цього ж металу, приміром, срібна дротинка у розчині AgNO3. Записують такий електрод як Ag│Ag+(AgNO3). Саме на поверхні розділу фаз, позначеній вертикальною рискою, відбувається реакція за участю електронів
Ag+ + e- ↔ Ag, яка визначає потенціал 0,058 [Ox] [Ag+]
Ex = E0 + ------ lg ------- = E0 + 0,058 lg----- = E0 + 0,058 lg[Ag+],
n [Red] [Ag]
при цьому треба враховувати, що, записуючи концентрацію, мають на увазі активність, а тверді та газоподібні речовини мають постійну концентрацію (як у насиченому розчині), що входить у Е0. Знайшовши Е0 для даної реакції у довіднику, можемо записати, що потенціал срібної дротинки, зануреної у розчин AgNO3 залежить від концентрації (активності) іонів аргентуму EAg = 0,7994 + 0,058 lg [Ag+], отже такий електрод можна використовувати як індикаторний для визначення іонів Ag+. Тільки малоактивні метали, що не реагують з водою і присутніми у розчині речовинами, не покриваються оксидною плівкою, можна використовувати як індикаторні електроди для визначення катіонів цього металу у розчині.
2)Електронообмінні електроди ІІ роду – це металева дротинка, вкрита малорозчинною сіллю цього металу і занурена у розчин солі з тим самим аніоном. Саме концентрація цього аніону визначає потенціал такого електроду. Приміром, срібна дротинка, на поверхні якої під час електрохімічної обробки утворена міцна плівка AgСІ. Записують такий електрод як Ag│AgСІ, КСІ. На поверхні розділу фаз, позначеній вертикальною рискою, відбувається реакція за участю електронів AgСІ + e- ↔ Ag + СІ–, EAg/AgCl=E0Ag/AgCl – 0,058 lg [Cl–].
Хлоридсрібний електрод можна використовувати як індикаторний для визначення хлорид-іонов, але найчастіше його використовують як електрод порівняння. Якщо заповнити електрод насиченим розчином КСІ, де концентрація хлорид-іонів залишається постійною, потенціал хлорид-срібного електроду залишається постійним під час вимірювань, що і потрібно для електрода порівняння. З досліджуваним розчином цей електрод сполучений електролітичним ключем у вигляді капіляру, через який розчин КСІ повільно витікає.
3)Окисно-відновні електроди – це платинова (або з іншого інертного металу) дротинка (пластинка), занурена у розчин, де існує рівновага між окисненою і відновленою формами певної речовини. Саме співвідношення концентрацій окисненої і відновленої форм у досліджуваному розчині визначають потенціал такого інертного електроду за рівнянням Нернста. Прикладом окисно-відновного електроду є хінгідронний електрод для вимірювання рН. Хінгідрон – це еквімолярна сполука хінону з гідрохіноном, яка у розчині створює обернену окисно-відновну пару: хінон С6Н4О2 + 2Н+ + 2 е- ↔ С6Н4(ОН)2 гідрохінон.
Якщо у цей розчин занурити платинову дротинку, її потенціал буде визначати логарифм концентрації (активності) іонів Н+, тобто рН:
0,058 [C6Н4О2]·[H+]2·
Ex = E0 + ------ lg ---------------= 0,699 + 0,058/2 lg [H+]2 =0,699 – 0,058рН
2 [C6Н4(ОН)2]
оскільки концентрації окисненої і відновленої форм рівні.
7.1.1.2.Іонообмінні електроди (які також називають мембранними, іон-селективними) – це такі електроди, на поверхні яких відбувається обмін іонами між досліджуваним розчином і мембраною, на поверхні якої і виникає потенціал. Його величина лінійно залежить від логарифму концентрації саме тих іонів, які переходять з розчину на поверхню мембрани. Реакція за участю електронів на поверхні електроду не відбувається.
1)Скляний електрод для вимірювання рН (рис.7.1). Це мембрана зі спеціального скла у формі кульки (1), що приєднана до скляної трубочки (3). Заповнений електрод 0,1 М розчином НСІ, у який занурена срібна дротинка(2), вкрита тонким шаром малорозчинної солі AgCI. Отже всередині знаходиться хлорид-срібний електрод порівняння, завдяки якому(4) скляний електрод можна підключити до приладу. За таким принципом побудовані усі мембранні електроди: іонообмінна мембрана контактує з двома розчинами, але внутрішній розчин залишається незмінним під час вимірювань, тому усі потенціали на внутрішніх поверхнях також не змінюються, а усі зміни потенціалу електроду пов’язані зі зміною складу зовнішнього досліджуваного розчину. Звичайне скло має загальну формулу Na2O·CaO·6SiO2. Отже це полімерний тримірний силікат, на поверхні якого іони Na+ замінюються на іони Н+ під час попереднього вимочування електроду в 0,1 М розчині НСІ: │ │
– Si – O– Na+ + H+ ↔ – Si – O– H+ + Na+
│ │
Н а
такій
гідратованій
поверхні
завдяки обміну іонів Н+
і виникає певний потенціал, що залежить
від концентрації (активності) цих іонів
а
такій
гідратованій
поверхні
завдяки обміну іонів Н+
і виникає певний потенціал, що залежить
від концентрації (активності) цих іонів
[Н+]внутр
E = К + 0,058 lg --------- = К + 0,058(рНзовн – рНвнутр.) = К′ + 0,058рН.
[Н+]зовн У величину К входить різниця потенціалів внутрішнього і зовнішнього електродів порівняння, потенціал рідинного з’єднання, потенціал асиметрії, зумовлений механічними напругами, що виникають у скляній мембрані. Величина К враховується на стадії калібровки, коли електроди занурюють у стандартний буферний розчин з точно відомим значенням рН і на шкалі реєструючого пристрою встановлюють це значення рН.
Скляний електрод поступово витіснив усі інші електроди, які також можна використати для вимірювання рН (водневий, хінгідронний, металоксидний). Він дозволяє вимірювати рН у досить агресивних середовищах, де інші електроди виходять з ладу. Але у сильнолужному середовищі при рН›9, де концентрація іонів Н+ різко зменшується, концентрація NaOH (точніше, іонів Na+) починає впливати на іонообмінну рівновагу на поверхні скляної мембрани (див.вище), виникає «лужна» помилка. Якщо замінити у складі скла Na2O на Li2O, можна виготовити мембрану, яка дозволяє вимірювати рН до 13. Рис.7.1.1.Скляний електрод.
2)Іонселективні електроди з рідинними мембранами – це такі електроди, де внутрішній розчин порівняння відокремлений від зовнішнього досліджуваного розчину пористою гідрофобною полімерною мембраною, заповненою органічною рідиною, у якій розчинена сіль потрібного нам іона з гідрофобним проти-іоном. Прикладом може бути нітратселективний електрод, чутливим елементом якого є плівкова мембрана, що складається з полівінілхлориду, диоктилфталату та нітрату тетраоктиламонію у співвідношенні 1:3:0,1.
Полівінілхлорид (–CH2–CHCl–)n – це твердий полімер, для пластифікації якого завжди використовували як пластифікатор рідину – диоктилфталат C6H4(COOC8H17)2. Якщо до 1 вагової частини твердого полімеру додають 3 частини гідрофобної рідини і розчиняють у ній 0,1 частину спорідненої до неї солі нітрату тетраоктил-амонію [(С8Н17)4N]+NO3–, отримують мембрану, селективну до нітрат-іонів. Катіон солі [(С8Н17)4N]+ гідрофобний – він не може перейти у водний розчин, а аніон NO3– – може. Завдяки цьому на поверхні мембрани і виникає потенціал, величина якого лінійно залежить від логарифму концентрації NO3–-іонів у розчині. Внутрішній розчин порівняння містить 0,1 М КNO3 і 0,1 М КСІ, у нього занурена срібна дротинка, вкрита AgCl, яка і підключає електрод до приладу. Потенціал на внутрішній поверхні мембрани, як і потенціал внутрішнього хлорид-срібного електроду порівняння, залишаються постійними, оскільки постійним залишається склад внутрішнього розчину. Отже потенціал такого електроду визначається концентрацією нітрат-іонів у зовнішньому досліджуваному розчині. Вимірювання можливі у межах від 1 М до 10-4 М концентрації нітратів. Для визначення менших концентрацій нітрату потрібні більш чутливі методи (молекулярна абсорбція, хроматографія та ін.)
7.1.1.3. Коефіцієнти селективності. Для усіх електродів у потенціометрії (на відміну від інших електрохімічних методів) характерна селективність: своїм потенціалом вони реагують на концентрацію тільки певних іонів у досліджуваному розчині і не реагують (в ідеальному випадку) на концентрацію усіх інших іонів у тому ж розчині. Сторонні електроліти, впливаючи на іонну силу розчину, визначають коефіцієнт активності і тільки таким чином повинні були б впливати на результати вимірювань, оскільки у рівняння Нернста входить саме активність – ефективна концентрація (концентрація помножена на коефіцієнт активності). Насправді ж сторонні іони впливають на іонообмінну рівновагу на поверхні іонообмінної мембрани, і, приміром, для скляного електроду (для вимірювання рН) у лужному середовищі, де зростає концентрація NaOH і КОН, залежність потенціалу від концентрації краще описується рівнянням Нернста-Айзенмана
E = const + (RT/F) ln([H+] + k1[Na+] + k2[K+] + …), де k1 і k2 – коефіцієнти селективності. Електрод для вимірювання рН повинен мати вкрай низькі величини k ( приблизно 10-3 і менше). Ці коефіцієнти не є константами, їх не можна використовувати для введення поправок, приміром, при вимірюванні рН. Але вони дають змогу оцінювати придатність різних електродів для вирішення конкретних аналітичних задач у присутності сторонніх іонів.
7.1.2. Пряма потенціометрія базується на вимірюванні потенціалу індикаторного електроду у серії стандартних розчинів з відомою концентрацією іонів, що визначаються, і вимірюванні його потенціалу у досліджуваному розчині у тих самих умовах. Оскільки за рівнянням Нернста залежність потенціалу лінійна не від концентрації, а від її логарифма, калібровка (аналог градуювального графіка) будується як залежність Е (потенціалу) від lgC. Приміром, для визначення нітратів в овочевій продукції, готують стандартні розчини з 10-1, 10-2, 10-3 і 10-4 М концентрацією KNO3, де –lg[NO3–] дорівнює 1; 2; 3 і 4. Стандартні розчини і суспензії зразків готують не на дистильованій воді, а на 1%-ному розчині KAl(SO4)2, який створює у цих розчинах однакову іонну силу. Для вимірювання потенціалу (аналітичного сигналу) у розчини занурюють індикаторний нітрат-селективний електрод і хлорид-срібний електрод порівняння, які підключають до потенціометра (іономіра И-160). Побудувавши калібровку і вимірявши потенціал у суспензії, приміром Е = 300 мВ, за графіком визначають, що у суспензії –lg[NO3–] = 3,4; тобто [NO3–] = 10-3,4 = 4·10-4 М.
Нітрати можна визначати у присутності сульфатів, хлоридів, гідрокарбонатів, ацетатів із коефіцієнтами селективності відповідно 0,001; 0,001; 0,002; 0,002.
Р ис.7.1.2.Калібровка
нітрат-селективного електроду
ис.7.1.2.Калібровка
нітрат-селективного електроду
7.1.3. Потенціометричне титрування – це вимірювання потенціалу індикаторного електроду під час титрування з метою побудови кривої титрування і графічного знаходження точки еквівалентності. Це один з електрохімічних способів встановлення точки еквівалентності, які використовують у тих випадках, коли кольорові індикатори не дають змоги її встановити, приміром, при титруванні темнозабарвлених розчинів або сумішей речовин, які треба визначити окремо. Таке потенціометричне титрування проводять дрібними порціями реактиву, зануривши у розчин електрод, індикаторний до іонів, концентрація яких у точці еквівалентності різко змінюється, і відповідний електрод порівняння.
Потенціометричне рН-титрування – це вимірювання рН під час титрування за допомогою скляного (індикаторного) та хлорид-срібного електродів, які занурюють у розчин і підключають до рН-метра. У випадку визначення кислот і сумішей кислот при поступовому додаванні порцій NaOH відбувається його реакція з кислотою, кислотність зменшується, а рН розчину збільшується, але нерівномірно – спочатку повільно, а потім – дуже різко. Цей стрибок рН ділять навпіл і знаходять точку еквівалентності. Побудувавши криву титрування – залежність рН від кількості доданого реактиву – і знайшовши точку еквівалентності, одержують кількість (об’єм) реактиву, що був витрачений на титрування кислоти Vт.екв. Існують різні способи знаходження точки еквівалентності при потенціометричному титруванні (див. рис.7.3) – інтегральна крива, диференційна крива титрування і крива титрування за другою похідною. Остання використовується в автоматичному титруванні, тому що у точці еквівалентності друга похідна змінює знак з (+) на (–) і прилад у цей момент припиняє додавання реактиву і фіксує кінець титрування.
7.2.Кондуктометрія(conductor–провідник). Метод базується на вимірюванні опору R (і розрахунку електропровідності W = 1/R) розчину залежно від концентрації речовини, яка у ньому визначається.
Як відомо, опір провідника R прямо пропорційний його довжині L і обернено пропорційний товщині S:
R = ρ·L/S, де ρ – питомий опір. На відміну від металевих провідників, де рухаються електрони, але у різному оточенні (різні метали мають різну кристалічну структуру), у розчині електроліту під дією електричного поля рухаються різні іони (що мають різну рухливість) в однаковому оточенні молекул розчинника. Електропровідність W розчину прямо пропорційна площині електродів S і обернено пропорційна відстані L між ними: W = χ·S/L, де χ – питома електропровідність. Вона залежить від концентрації С (моль-екв/л) електроліту у розчині: χ = λ· С/1000 (1000 мл в 1 л), де λ – еквівалентна електропровідність. Ця величина не повинна була б залежати від концентрації, але на неї впливає іонна сила розчину, яку визначає концентрація усіх електролітів. Зменшення концентрації зменшує іонну силу, міжіонна взаємодія зменшується і λ зростає, прагнучи певного значення. Це еквівалентна електропровідність за нескінченного розведення, яка складається з двох доданків – рухливостей катіона і аніона λ∞ = λ(+) + λ(–).
Нещодавно введено уточнення, яке розрізняє рухливість “u” і еквівалентну електропровідність λ на число Фарадея F: λ(+) = F·u(+); λ(–) = F·u(–). Але для спрощення часто λ(–) і λ(+) називають рухливостями. Ці величини визначені для багатьох катіонів і аніонів, знаходяться у довіднику і дають змогу розрахувати електропровідність різних електролітів.
7.2.1.Пряма кондуктометрія В об’єктах харчової та біотехнології пряма кондуктометрія дає змогу визначати електроліти у присутності не-електролітів. У технологічному розчині якісний склад відомий, тому можна побудувати залежність електропровідності від концентрації електроліту і визначати його концентрацію за методом градуювального графіка. Але випадкова присутність будь-якого стороннього електроліту робить таке визначення неможливим. Основна відмінність кондуктометрії від потенціометрії – це неселективність кондуктометрії. Електроди у кондуктометричних вимірюваннях реагують на усі присутні у досліджуваному розчині електроліти, що створюють електропровідність, не розрізняючи їх.
7.2.2.Кондуктометричне титрування. Один електроліт у присутності інших (теж електролітів) можна визначити, якщо підібрати реактив, який буде реагувати тільки з даним електролітом і провести кондуктометричне титрування. Це вимірювання опору під час титрування з метою побудови кривої титрування і графічного знаходження точки еквівалентності. Як приклад можна навести кондуктометричне титрування двох речовин в одному розчині. Для визначення загального вмісту білку пробу харчового продукту розчиняють у концентрованій H2SO4 – при цьому відбувається так звана «мокра мінералізація»: білки, жири і вуглеводи руйнуються і перетворюються на Н2О і СО2, але аміногрупи, що входять до складу білків, дають ще іони амонію. Визначивши іони амонію, можна оцінити загальний вміст білкового нітрогену у даному харчовому продукті. Як відомо, пряме титрування іонів амонію розчином лугу з кольоровими індикаторами неможливе. Використовуючи кондуктометричне титрування можна прямо відтитрувати іони амонію у присутності залишку сульфатної кислоти. Послідовність перетворень у молекулярному і іонному вигляді має такий вигляд: H2SO4 + 2 NaOH = Na2SO4 + 2 H2O;
2H++SO42-+2Na++2OH- =2Na++SO42-+2H2O; K(H2O)=10-14;
(NH4)2SO4 + 2 NaOH = Na2SO4 + 2 NH4OH;
2NH4++SO42-+2Na++OH- =2Na++SO42-+2NH4OH; K(NH4OH)=2·10-5;
Утворення слабкого електроліту є рушійною силою іонної реакції. Менша константа дисоціації відповідає сильнішому зв’язку між іонами, тому першою відбувається реакція утворення Н2О. Тільки коли вона пройде на 99,99%, почнеться реакція утворення NH4OH. Отже, при поступовому додаванні порцій (0,3 мл) реактиву NaOH він не з’являється між електродами, а перетворюється у першій реакції. Внаслідок цієї реакції кількість іонів між електродами не змінюється, але іони Н+ (з рухливістю 362) замінюються на іони Na+ (з рухливістю 52). Електропровідність зменшується, опір збільшується. Після першої точки еквівалентності починається друга реакція, внаслідок якої кількість іонів між електродами також не змінюється, але іони NH4+ (з рухливістю 76) замінюються на іони Na+ (з рухливістю 52). Електропровідність зменшується, опір зростає (але ледь помітно). Після другої точки еквівалентності, коли обидві речовини відтитровані, між електродами з’являється надлишок реактиву: іони Na+ (52) і ОН- (205). Електропровідність збільшується, опір зменшується. Цим трьом процесам, які відбуваються під час титрування, на кривій титрування відповідають три лінії і дві точки їхнього перетинання, що співпадають з точками еквівалентності.
Побудувавши криву титрування і знайшовши точки еквівалентності, одержують кількість (об’єм) реактиву, що був витрачений на титрування першої речовини V1 і другої – (V2 – V1). Якщо треба розрахувати кількість кислоти, підставляють її еквівалент Mr(½H2SO4) =98/2 =49, а для розрахунку білкового азоту Mr(N) = 14.
7.2.3.Електроди у кондуктометрії. Вимірювання опору у розчинах електролітів проводять при певній частоті змінного електричного струму для уникнення небажаних процесів на поверхні електродів (якщо струм постійний, відбувається електроліз). Ці процеси змінюють поверхню і заважають точним вимірюванням.
Низькочастотна кондуктометрія використовує струм з частотою від 1000 до 10000 Гц, а високочастотна – з частою у десятки мегагерц. В останньому випадку електроди не треба занурювати у розчин – вони охоплюють посуд із досліджуваним розчином. Це зручно для контролю продукції у запаяних скляних ампулах. Отже, низькочастотна кондуктометрія є контактною, а високочастотна – безконтактною. Високочастотні (безконтактні) вимірювання мають певні переваги. Не потрібні інертні дорогоцінні метали для виготовлення електродів. Електроди не занурюють в агресивне середовище під час вимірювань, вони не впливають на хімічний склад розчину. У високочастотних титраторах можна використовувати реакції з утворенням осадів, які б осідали на звичайних електродах і заважали б титруванню.
7.3. Електроаналіз – електрогравіметрія та кулонометрія. Електроліз – це окисно-відновна реакція, що відбувається на поверхні електродів, занурених у розчин електроліту, якщо крізь цей розчин пропускають сталий електричний струм. На катоді катіони приєднують електрони – відновлюються, а на аноді аніони віддають електрони – окиснюються. Ця реакція широко використовується у багатьох технологічних процесах хімічного виробництва важливих речовин: водню, кисню, хлору, гідроксидів натрію, калію та ін. Проте за певних умов електроліз можна використати як аналітичну реакцію для кількісного визначення тих речовин, які містяться у розчині і здатні брати участь в електрохімічних перетвореннях на поверхні електродів. Реактивом у цьому разі є електричний струм (електрони). Як і у будь-якій аналітичній реакції тут можна вимірювати кількість продукту, г, що відкладається (осаджується) на одному з електродів, або кількість реактиву – електрики, що вимірюється в кулонах. Перший варіант називають електрогравіметрією, а другий – кулонометрією.
7.3.1. Електрогравіметрія (електроваговий аналіз) базується на тому ж принципі, що і хімічний ваговий аналіз (гравіметрія). Речовина, яку треба визначити у розчині, бере участь в електрохімічній реакції і осаджується на поверхні одного з електродів. На катоді можуть осаджуватися метали: мідь, нікель, цинк, кадмій та ін. На аноді осаджується свинець у вигляді PbO2. Виходячи з таблиці стандартних потенціалів і рівняння Нернста, розраховують і подають на електроди напругу, достатню для кількісного виділення, приміром, міді, і недостатню для виділення цинку або інших металів, які забруднювали б осад. Густина струму на поверхні електроду має забезпечувати утворення щільного осаду, що не осипається під час промивання, висушування і зважування електроду з осадом. Кількість речовини розраховують за результатами зважування електроду до і після електролізу. Метод використовують для визначення металів, що входять до складу сплавів, з яких виготовляють апаратуру харчових виробництв і упаковку для харчових продуктів.
7.3.2. Кулонометрія. Вимірявши кількість електрики (в кулонах), що витрачається на електрохімічне перетворення на поверхні електродів, можна розрахувати масу речовини, г, яка бере участь у цьому перетворенні за формулою, що відповідає законам Фарадея:
m
=
![]()
![]() I
t
=
Q,
I
t
=
Q,
де F – число Фарадея – кількість кулонів, що витрачається на перетворення 1 моль-екв речовини (96487 К), Mr – молекулярна або атомна маса речовини, що перетворюється; n – кількість електронів, що бере участь в електрохімічній реакції на поверхні електрода; I – сила струму, а; t – тривалість електролізу, с; Q – кількість електрики, К. За цим рівнянням можна оцінити чутливість кулонометричного методу. Якщо, приміром, відновлювати іони Ag+ протягом 20 хв струмом 10–6 а, можна визначити 1 мкг (10–6 г) цього металу. Зараз можна вимірювати з достатньою точністю силу струму 10–7 – 10–8 а і час в кілька секунд. Тому чутливість методу – найменша кількість речовини, яку можна визначати кулонометрично, становить соті й тисячні частки мікрограма. Отже, кулонометрію використовують для визначення мікроелементів у харчових продуктах, зокрема мікрокількостей забрудників – важких металів. Кулонометрія використовує значно більшу кількість електрохімічних реакцій, ніж електрогравіметрія. Їх можна розділити на такі групи.
Перша група – відновлення іонів металів і виділення їх у вільному стані: Men+ + n e- = Me0.
Таким способом визначають Cu, Pb, Cd, Bi та інші метали. Зручним катодом є ртуть, оскільки утворення амальгам полегшує виділення багатьох металів: Men+ + n e– + Hg = MeHg,
а виділення водню утруднюється високою перенапругою на поверхні ртуті. Цим методом можна аналізувати і суміш катіонів кількох металів, виділяючи з розчину електролізом спочатку більш електропозитивні елементи, а потім – більш електронегативні (якщо різниця потенціалів виділення становить 0,3 В і більше).
Друга група визначень використовує зворотний процес – анодне окиснення металів, попередньо виділених електролізом з досліджуваного розчину: Me0 = Men+ + n e–.
Часто цей варіант використовують для аналізу металевих покриттів і оксидних плівок, якщо вони розчиняються під час анодного процесу, а основа – ні.
Третя група визначень – це електрохімічне перетворення, вихідні речовини і продукти якого залишаються у розчині: Men+ + (n – m) e– = Mem+.
Усі наведені варіанти об’єднують назвою пряма кулонометрія. Значно більше можливостей для визначення надає кулонометричне титрування. Цей метод використовує електрохімічну реакцію для утворення у розчині реактиву, який потім кількісно реагує з речовиною, що визначається.
Прикладом кулонометричного титрування може бути визначення феруму(ІІ). Класичним методом ферум(ІІ) визначають титруванням окисниками, серед яких є і церий(IV): Fe2+ + Ce4+ = Fe3+ + Ce3+,
Прямою кулонометрією ферум(ІІ) можна окиснювати на аноді: Fe2+ – e– = Fe3+,
але при зменшенні концентрації феруму(ІІ) починається побічна реакція електролізу води, на яку витрачається струм. Якщо ж створити у розчині достатню концентрацію церію(ІІІ), струм витрачається на реакцію утворення церію (IV): Ce3+ – e– = Ce4+,
який окиснює залишок феруму (ІІ), як у класичному титруванні: Fe2+ + Ce4+ = Fe3+ + Ce3+,
тобто уся кількість електрики, що вимірюється, витрачається у кінцевому результаті на окиснення феруму. Кінець титрування визначають потенціометрично, занурюючи у розчин платиновий індикаторний електрод. Його потенціал, що залежить від співвідношення концентрацій Fe3+ і Fe2+:
E = 0,77 + 0,058 lg Fe3+/Fe2+, різко збільшується у точці еквівалентності.
Отже, розробляючи методику кулонометричного визначення потрібно:
1) створити такі умови проведення електролізу, щоб струм витрачався тільки на одну реакцію і не витрачався на будь-які побічні реакції («вихід за струмом» був 100 %);
2) мати спосіб визначення моменту закінчення цієї реакції;
3) точно визначити кількість електрики, що була витрачена на цю реакцію.
Метод кулонометричного титрування за сталої сили струму відкриває надзвичайні можливості точного визначення малих кількостей біологічно активних речовин з використанням усіх типів аналітичних реакцій: кислотно-основних, окисно-відновних, утворення комплексних сполук та осадження.
Метод грунтується на тому, що кількість електрики визначають хронометрично: протягом усього терміну електролізу силу струму підтримують незмінною і тривалість процесу точно вимірюють (секундоміром або більш точними електронними приладами). Кількість електрики розраховують, помноживши силу струму на час: Q = It.
Для підтримування незмінної сили струму послідовно з електролітичною коміркою підключають опір 10000 – 25000 ом, при цьому напруга джерела струму становить 100 – 200 В. Напруга на електродах комірки може підвищуватись під час електролізу зі зменшенням концентрації іонів у розчині, але це підвищення рідко перевищує кілька десятих вольта. У порівнянні з величиною напруги джерела струму воно незначне, тому сила струму під час електролізу залишається практично незмінною. Існує апаратура, що дозволяє автоматично підтримувати силу струму з відхиленнями 0,1%, а тривалість електролізу визначати з точністю до 0,1%.
7.4.Вольтамперометрія (полярографія та амперометричне титрування) використовує явище концентраційної поляризації одного з двох електродів у досліджуваному розчині залежно від напруги, що подається на ці електроди під час електролізу. Один з двох електродів (мікроелектрод) має поверхню у 100 раз меншу ніж інший, тому (завдяки високій густині струму) на ньому відбувається концентраційна поляризація і спостерігається явище граничного дифузійного струму. Величина цього струму прямо пропорційна концентрації електроактивної речовини у досліджуваному розчині.
У 1922-1925 роках чеський хімік Я.Гейровський, використовуючи ртутний крапельний електрод, розробив метод визначення хімічного складу розчинів, який він назвав полярографією. А через 30 років за цю розробку йому присудили Нобелівську премію з хімії.
Крім ртутного крапельного електроду використовують мікроелектроди з Pt, Au, амальгамований срібний Ag(Hg) та інші. У цьому випадку метод називають вольтамперометрією, залишивши назву «полярографія» для ртутного крапельного електроду.
7.4.1.Полярографічна хвиля. Залежність сили струму, що проходить через електролітичну комірку (І,мкА), від напруги (Е,В), що подається на неї (див.рис.7.4.1.) називають полярографічною хвилею (вольтамперометрична – вольти подаються на комірку, ампери реєструються). На початку хвилі (АБ) спостерігається невеликкий залишковий струм. Він пов’язаний з відновленням залишків розчиненого кисню (О2), який треба видаляти продуванням розчину азотом (N2). Крім того цей струм пов’язаний з утворенням на поверхні мікроелектроду своєрідного конденсатора з тих іонів, які цого оточують. Зі збільшенням від’ємного потенціалу мікрокатоду у точці Б при потенціалі виділення починається процес відновлення іонів металу Pb2++2e-→Pb(Hg). Подальше збільшення струму (БВ) пов’язане зі збільшенням кількості іонів, що відновлюються, в одиницю часу. Якісною характеристикою іонів, що відновлюються, є потенціал напівхвилі Е½ , тобто потенціал, який відповідає Ід/2, тому що саме цей потенціал не залежить від концентрації.
Коли усі іони, що можуть потрапити до поверхні мікроелектроду за рахунок дифузії, відновлюються, струм досягає граничного значення (ВГ) і називається граничним дифузійним струмом. Дифузія пропорційна градієнту концентрації, а концентрація іонів, що відновлюються, у приелектродному шарі стає нульовою. Отже граничний дифузійний струм пропорційний концентрації іонів у розчині і він може бути аналітичним сигналом для визначення цих іонів. Іd=kС, де С – концентрація іонів, що визначаються; k – коефіцієнт пропорційності.
При подальшому збільшенні від’ємного потенціалу мікрокатоду (Г) на полярограмі з’явиться декілька хвиль, якщо в аналізованому розчині присутні декілька речовин, які відновлюються на мікрокатоді за різних потенціалів. За величиною потенціалу кожної півхвилі визначають, який компонент, а за силою дифузійного струму – концентрацію цього компоненту.
Якщо окремі хвилі важко розрізнити на звичайній полярограмі, краще побудувати диференційну (рис.7.4.1), тобто залежність першої похідної збільшення струму від збільшення потенціалу мікроелектроду – так званий «полярографічний спектр». Кожному компоненту, що відновлюється у певній послідовності, на ньому відповідає максимум. Положення максимуму (Е½) вказує, який компонент відновлюється, а висота h пропорційна його концентрації у розчині.
7.4.2.Полярографічний фон. Iони до поверхні електроду можуть потрапляти не тільки завдяки дифузії, але і під дією самого електричного поля («міграція іонів»). Цей додатковий «міграційний струм» порушує пропорційність струму, що реєструється, концентрації речовини, яка визначається.
Для уникнення «міграційного струму» у розчин додають великий надлишок сторонніх електролітів (полярографічний фон), іони яких не відновлюються в умовах аналізу (тому що для їхнього відновлення потрібні значно більш від'ємні потенціали, ніж для визначуваних іонів), а тому не заважають полярографуванню. Під дією електричного поля ці іони тільки рухаються до мікроелектроду і «екранують» його (закривають екраном) для тих іонів, які визначаються. Як індиферентні (в умовах аналізу) електроліти використовують в основному солі KCI, KNO3, NaCl, NH4Cl, LiClО4 тощо.
7.4.4.Амперометричне титрування – це метод об’ємного аналізу (титриметрії), у якому для встановлення точки еквівалентності використовують полярографічний пристрій. У розчин під час його титрування занурюють два електроди – мікроелектрод і електрод порівняння. На електроди подають напругу, яка відповідає граничному дифузійному струму (рис.7.4.1) речовини, яка визначається, або реактиву, яким титрують розчин. Силу струму (Ід) реєструють у процесі титрування і будують графік залежності Ід від кількості (V) доданого реактиву – криву титрування. Якщо титрують, приміром, іони Pb2+ сульфатом, відбувається реакція Pb2+ + SO42- = PbSO4↓, внаслідок якої концентрація Pb2+ і його дифузійний струм рівномірно зменшується до точки еквівалентності. Після точки еквівалентності Ід вкрай незначний – у розчині відповідно добутку розчинності залишається [Pb2+]≤10-4M (3.4.4), а сульфат не відновлюється на мікроелектроді (рис7.4.2). Об’єм реактиву у точці еквівалентності знаходять як проекцію точки перетинання двох прямих частин кривої титрування.
Частіше розчином нітрату плюмбуму Pb(NO3)2 титрують сульфати, карбонати, молібдати, оксалати та інші аніони, які не здатні відновлюватись на мікроелектроді і давати струм. У цьому випадку струм зростає після точки еквівалентності, коли у розчині з’являється надлишок іонів Pb2+, які легко відновлюються (рис.7.4.3). Якщо ж розчином Pb(NO3)2 титрують хромат CrO42-,який дає струм до точки еквівалентності, отримують третій варіант кривої амперометричного титрування. До точки еквівалентності струм зменшується, оскільки зменшується концентрація хромату. Після точки еквівалентності додається надлишок Pb2+, які збільшують струм (рис.7.4.4).
7.4.5.Біамперометричне титрування – це титрування з двома інертними (платиновими) електродами. Якщо на електроди подається невелика напруга, струм можна буде зафіксувати тільки у тому випадку, коли на катоді окиснена форма відновлюється, а на аноді відновлена форма окиснюється. Такі системи називають оберненими. Приміром, титрування Fe2+ до Fe3+ окисниками неодноразово розглядалось раніше. На початку титрування на

 Рис.7.4.5
Рис.7.4.5
аноді може окиснюватись Fe2+ = е- + Fe3+, але на катоді можлива реакція Fe3+ + е- =Fe2+ не відбувається, тому що Fe3+ поки що немає. У процесі титрування концентрація Fe2+ зменшується, а Fe3+ збільшується і струм зростає за рахунок реакції на катоді Fe3+ + е- = Fe2+. Він стає максимальним, коли 50% Fe2+ буде відтитровано до Fe3+, їхні концентрації стануть однаковими: окиснена форма забезпечує реакцію на катоді, відновлена – на аноді. Після цього струм буде обмежувати реакція на аноді Fe2+ = е- + Fe3+, оскільки концентрація Fe2+ поступово зменшується до точки еквівалентності і струм зменшується до нуля у точці еквівалентності. Далі усе залежатиме від реактиву, надлишок якого з’являється у розчині після точки еквівалентності. Якщо окиснена і відновлена форми реактиву складають не обернену систему, струм зростати не буде, якщо обернену – струм буде зростати після точки еквівалентності (рис.7.4.5).
Прикладом титрування не оберненої системи оберненою (рис 7.4.6) є визначення амонійного (білкового) нітрогену гіпобромітом: (NH4)2SO4 + 3 NaBrO + 2 NaOH= N2+ 3 NaBr+ Na2SO4+ 5H2O.
 У
ході титрування гіпоброміт витрачається
на окиснення іонів амонію до молякулярного
азоту. Реакція необоротна і стехіометрична
за певних умов: а)створення слабколужного
середовища (рН 8-9) додаванням 0,1 М розчину
тетраборату натрію; б)для створення
достатньої електропровідності розчину
перед титруванням у пробу додають 35 % -
ний розчин NaBr.
Кількість амонійного нітрогену множимо
на коефіцієнт перерахунку амонійного
нітрогену на білок (для різних продуктів
у довідниках наводяться різні значення)
6,25 і отримуємо вміст білків у пробі.
У
ході титрування гіпоброміт витрачається
на окиснення іонів амонію до молякулярного
азоту. Реакція необоротна і стехіометрична
за певних умов: а)створення слабколужного
середовища (рН 8-9) додаванням 0,1 М розчину
тетраборату натрію; б)для створення
достатньої електропровідності розчину
перед титруванням у пробу додають 35 % -
ний розчин NaBr.
Кількість амонійного нітрогену множимо
на коефіцієнт перерахунку амонійного
нітрогену на білок (для різних продуктів
у довідниках наводяться різні значення)
6,25 і отримуємо вміст білків у пробі.
Рис.7.4.6
7.4.6. Удосконалення методів вольтамперометрії за рахунок різних електротехнічних та електронних пристроїв призвело до появи високошвидкісної полярографії, циклічної вольтамперометрії, диференційної імпульсної полярографії, інверсійної анодної вольтамперометрії та хронопотенціометрії. Останні методи дають змогу визначати вкрай незначні (10-7–10-10 М) концентрації важких металів (токсичних елементів) у продуктах харчування, об’єктах біотехнології та довкілля.