

Astruc D. - Modern arene chemistry (2002)(en)
.pdf4.7 Mechanism of Aryl Halide Amination and Etheration 153
Scheme 4. Reductive elimination of arylamines from PPh3-ligated PdII amido complexes.
The best-understood examples of reductive eliminations that form the CaN bond in amines involve palladium complexes. Both Boncella et al. [66] and Hartwig et al. [63, 64, 101] have observed these reactions with palladium amido aryl complexes. Hartwig’s group has studied the mechanism of this process in detail [101, 280]. Although monomeric and dimeric amido complexes have been isolated, the monomeric species undergoes reductive elimination. For complexes with monodentate ligands, kinetic studies have indicated that the actual CaN bond formation occurs simultaneously from both threeand four-coordinate intermediates. With chelating phosphines, therefore, the chemistry is likely to occur directly from the four-coordinate complexes observed in solution. The reductive elimination of arylamines is favored by increasing nucleophilicity of the amido group and by increasing electrophilicity of the aryl group.
The mechanisms of the reductive eliminations shown in Scheme 4 have been studied [101, 280]. Potential pathways for reductive elimination are shown in Scheme 5. The reduc-
Scheme 5. Potential mechanisms for reductive elimination of arylamines from PPh3-ligated PdII amido complexes.

1544 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
tive eliminations from the monomeric diarylamido aryl complex 23 illustrate two important points concerning the elimination reactions. First, these reactions were found to be first order, demonstrating that the actual CaN bond formation occurred from a monomeric complex. Second, the observed rate constant for the elimination reaction contained two terms (Eq. (51)), corresponding to the two concurrent mechanisms, Path B and Path C in Scheme 5. One of these mechanisms involves initial, reversible phosphine dissociation followed by CaN bond formation from the resulting 14-electron, three-coordinate intermediate. The second mechanism involves reductive elimination from a 16-electron, four-coordinate intermediate, presumably after trans-to-cis isomerization. The dimeric amido complexes undergo reductive elimination after cleavage to form two monomeric, three-coordinate, 14electron amido complexes.
d½22&=dt ¼ kobs½22& |
ð51Þ |
||
kobs ¼ K2k2 þ |
K3k3 |
|
|
½PPh3& |
|
||
The observation that the reductive elimination process involves a pathway proceeding via four-coordinate, presumably cis, monomeric amido aryl complexes led to the preparation of palladium amido complexes with chelating ligands [64]. Results with these complexes confirmed this conclusion and led to the development in our laboratory of second-generation catalysts based on palladium complexes with chelating ligands [281]. The DPPF-ligated palladium amido aryl complexes in Scheme 6 were found to undergo reductive elimination of arylamines in high yields [64, 101]. The rates of these reactions were first order in palladium and zero order in the trapping ligand. Thus, the data on the reductive elimination reactions are consistent with a direct, concerted formation of the CaN bond from the cis, fourcoordinate DPPF complex.
Because the reductive elimination from DPPF-ligated palladium does not involve geometric rearrangements or changes in coordination number before the rate-determining step, the DPPF complexes allowed an assessment to be made of the electronic properties of the transition state in this reaction. The relative rates for elimination from amido groups were found
Scheme 6. Reductive elimination of arylamines from DPPF-ligated palladium amido complexes.
4.7 Mechanism of Aryl Halide Amination and Etheration 155
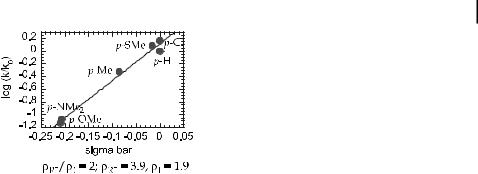
Fig. 4. Electronic analysis using a combination of sI, and sR0 for the reductive elimination of aryl arylamines.
to decrease in the order alkylamido > arylamido > diarylamido. This trend implies that the more nucleophilic the amido group, the faster the elimination process. Variation of the aryl group led to similar results as those seen in an extensive study of the electronic aspects of CaS bond-forming eliminations of sulfides [54]. The data for electronic e ects on sulfide and amine eliminations (Figure 4) show that electron-withdrawing groups accelerate the reductive elimination process and that substituents with large sR values a ect the reaction rates more than substituents with large sI values. Resonance e ects are stronger than inductive e ects, perhaps due to arene coordination during the reaction. In a more rough sense, the amido group acts as a nucleophile and the aryl group as the electrophile.
Although it focused on the reductive elimination of ethers and not amines, a recent study revealed the importance of steric hindrance in accelerating the reductive elimination process. Until recently, reductive elimination of acyclic aryl ethers had only been achieved from palladium complexes containing highly electron-poor palladium-bound aryl groups [65]. The lower nucleophilicity of alkoxides and aryl oxides, relative to amides, makes the elimination process su ciently slow that reactions occur only with palladium complexes bearing strongly electrophilic aryl groups. However, the use of ligands with demanding steric properties was found to accelerate this reaction to the point that reductive elimination was observed from complexes containing electron-neutral aryl groups bound to palladium [282]. The dimeric phenoxide complex fPd[FcP(tBu)2](o-tolyl)(OC6H4-4-OMe)}2 gave diaryl ether at 70 C in roughly 20 % yield. Addition of P(tBu)3 to this complex led to some phosphine exchange that ultimately provided the diaryl ether in close to quantitative yield. Thus, the strong electron-donating property of alkylphosphines is dominated by the steric demands, and this steric demand appears to have a large accelerating a ect on the rate of reductive elimination.
4.7.4
Competing b-Hydrogen Elimination from Amido Complexes
The amination chemistry depends on preventing irreversible b-hydrogen elimination from the amido complexes before reductive elimination of the amine. At the early stages of the development of the amination chemistry, it was remarkable that the unknown reductive elimination of arylamines could be faster than the presumed rapid [71, 72] b-hydrogen

1564 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
elimination from late transition metal amides. In fact, directly observed b-hydrogen elimination from late transition metal amido complexes was rare ADDIN and there were no known examples of its irreversible occurrence from a simple monomeric amido species [283].
ð52Þ
Recently, Hartwig prepared 16-electron, square-planar amido complexes that undergo irreversible b-hydrogen elimination [102]. This observation allowed the beginning of a mechanistic understanding of this process and highlighted the unfounded assumption that this b- hydrogen elimination process is typically rapid. The N-benzyl anilide 25 in Eq. (52) produced the stable imine and iridium hydride 26 in nearly quantitative yields. b-Hydrogen elimination from 25 required temperatures of 100 C or above, while elimination from an alkylamide required 70 C. In contrast, Schwartz showed that the analogous alkyl complexes underwent b-hydrogen elimination below room temperature [284, 285].
ð53Þ
The intermediate that underwent CaH bond cleavage was a 14-electron, three-coordinate complex that formed by reversible phosphine dissociation (Eq. (53)). Importantly, there was no detectable competing b-hydrogen elimination from a 16-electron, four-coordinate complex. The mechanism for b-hydrogen elimination from a 14-electron intermediate parallels that for b-hydrogen elimination from square-planar alkyl complexes [286, 287]. b-Hydrogen elimination from analogous alkoxides has been studied by Zhao, Hesslink, and Hartwig [288]. b-Hydrogen elimination from the alkoxide complexes occurs at similar rates to that from the amides. Detailed mechanistic studies have shown that alkoxide b-hydrogen elimination is reversible and that the irreversible step is the displacement of the resulting aldehyde or ketone from the metal center by free phosphine.
4.7.5
Selectivity : Reductive Elimination vs. b-Hydrogen Elimination
Two studies have been conducted that outline the e ects of the steric and electronic properties of the ligand on the relative rates for reductive elimination of amine and b-hydrogen elimination from amides. One study focused on the amination chemistry catalyzed by P(o- C6H4Me)3 palladium complexes [135], while the second focused on the chemistry catalyzed by complexes containing chelating ligands [106].
Studies of aryl halide amination with secondary aminostannanes and palladium catalysts bearing P(o-C6H4Me)3 ligands are summarized in Scheme 7. These studies revealed four
4.7 Mechanism of Aryl Halide Amination and Etheration 157
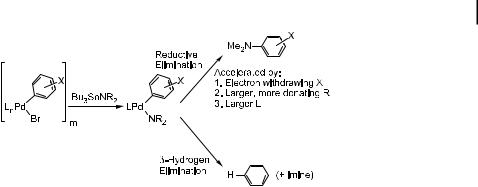
Scheme 7. Factors controlling selectivity for amination vs. hydrodehalogenation of aryl halides.
factors that control the amount of product formed from aryl halide hydrodehalogenation vs. the amount formed by amination. First, aryl halides bearing electron-withdrawing groups on the aryl ring gave more amination product and less hydrodehalogenation product than did those bearing electron-donating groups. This result is consistent with the faster reductive elimination of amines from complexes with electron-poor aromatic groups discussed above. Second, reactions of N-alkyl arylamides gave more hydrodehalogenation product than did those of dialkylamides, consistent with reductive elimination from complexes of arylamides being slower than that from complexes of dialkylamides. Third, deuterium-labeling experiments showed that the majority of the dehalogenation product after catalyst initiation came from b-hydrogen elimination from the amido group.
The final point concerns the steric e ects of the phosphine aryl groups on the relative amounts of arene and arylamine products. Careful monitoring of the products formed from both stoichiometric and catalytic reactions of palladium complexes containing P(o- C6H4Me)3, P(o-C6H4Me)2Ph, P(o-C6H4Me)Ph2, and PPh3 showed steadily decreasing ratios of amine to arene as the size of the ligand was decreased. Thus, larger phosphine ligands enhance the rate of reductive elimination of amines relative to the rate of b-hydrogen elimination. Reductive amine elimination decreases the coordination number of the metal, whereas b-hydrogen elimination from an amide either increases the coordination number of the metal because of the formation of a coordinated imine along with the hydride, or leaves it unchanged if imine is extruded without coordination. Large groups on the phosphine ligand enhance the rates of reactions that decrease the coordination number, and will, therefore, increase the rate of reductive elimination [289] of amines relative to the rate of b- hydrogen elimination. This study foreshadowed the ability of even larger tert-butylphosphine ligands to drive the amido intermediate toward reductive elimination to form the arylamine instead of undergoing b-hydrogen elimination, even when sterically undemanding primary amines are used as substrates [164, 290].
Results obtained with chelating ligands displaying varied steric properties contrasted those obtained with monodentate ligands [106]. Large, chelating phosphine ligands such as bis- 1,10-(di-o-tolylphosphino)ferrocene gave more hydrodehalogenation product than did DPPF. Reactions catalyzed by complexes of electron-poor DPPF derivatives, which should generate a more electron-poor metal and favor reductive elimination of amine, gave more arene than did those catalyzed by complexes of electron-rich DPPF derivatives. Further, ligands with large bite angles gave more arene than those with small bite angles, in contrast to what

1584 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
would be expected from previous studies that showed an increase in the rate of CaC bondforming reductive elimination with increasing bite angle [291]. Surprisingly, the arene product produced by these substrates contained no deuterium when formed from reactions catalyzed by complexes of many chelating phosphines. The source of hydrogen remains unclear, but these results demonstrated that much of the arene generated in reactions conducted with DPPF or BINAP as ligand does not form by a simple b-hydrogen elimination and CaH bond-forming reductive elimination sequence.
4.7.6
Overall Catalytic Cycle with Specific Intermediates
At this time, one can combine the results on reductive elimination and oxidative addition to construct a mechanism for the amination chemistry catalyzed by palladium complexes containing several types of both monodentate and chelating ligands. These catalytic cycles di er in the coordination number and charge of the palladium complexes that lie on the catalytic cycle, the factors that control amination vs. aryl halide reduction, and the role of base in inducing rapid oxidative addition. It has been shown that the catalytic cycle for the amination of aryl halides catalyzed by P(o-C6H4Me)3 involves exclusively intermediates with a single phosphine ligand. In contrast, the chemistry catalyzed by DPPF or BINAP palladium complexes involves bis(phosphine) complexes as a result of ligand chelation
4.7.6.1 Mechanism for Amination Catalyzed by P(o-C6H4Me)3 Palladium Complexes
Scheme 8 shows an experimentally supported mechanism for amination catalyzed by P(o- C6H4Me)3 phosphine complexes. The Pd0 complex is a 14-electron, two-coordinate species, from which one of the phosphine ligands dissociates before aryl halide oxidative addition. The aryl halide complexes react with an amine to generate amine-ligated aryl halide complexes either by reaction with the monomer or by an associative reaction with dimeric 4. Reactions of these amine complexes with base generate three-coordinate amido species, which undergo rapid reductive elimination. In the case of reactions catalyzed by complexes containing monodentate ligands, large phosphines accelerate the overall rate by favoring monophosphine complexes and provide good selectivity by accelerating reductive elimination relative to b-hydrogen elimination.
Scheme 8. Overall mechanism for aryl halide amination catalyzed by
P(o-C6H4Me)3-ligated palladium complexes.
4.7 Mechanism of Aryl Halide Amination and Etheration 159
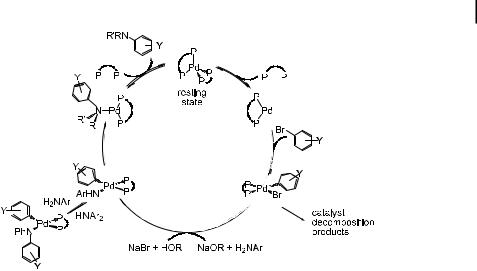
Scheme 9. Overall mechanism for aryl halide amination catalyzed by bis(phosphine)-ligated palladium complexes.
4.7.6.2 Mechanism for Amination Catalyzed by Palladium Complexes with Chelating Ligands
Scheme 9 shows a mechanism for the amination of aryl halides catalyzed by DPPFor BINAP-ligated palladium complexes, and a similar mechanism is presumably followed by reactions catalyzed by structurally related bis(phosphine) complexes. As discussed in the section on oxidative addition of aryl halides, the monochelate Pd(0) complex is formed by dissociation of BINAP or DPPF, and these 14-electron intermediates add aryl halide. The monochelate complex containing DPPF as the ligand undergoes recoordination of phosphine faster than oxidative addition of aryl halide, while the monochelate complex containing BINAP adds aryl bromide faster than it recoordinates ligand when an excess of aryl bromide is present. The amido complex is generated by either deprotonation of coordinated amine or by reaction of amine with an intermediate alkoxide complex [65]. When aryl sulfonates are used as substrates, a third pathway for amide generation is possible. The triflate ligand can be displaced by amine to generate a cationic amine complex. The coordinated amine in this species would then be deprotonated by even weak phosphate or carbonate base.
The resulting amido aryl complexes undergo reductive elimination directly from the 16-electron, four-coordinate complex, rather than from the three-coordinate, 14-electron, monophosphine complex generated when catalysts bearing sterically hindered monophosphines are used. For reactions involving chelating phosphines, the selectivity for reductive elimination, rather than b-hydrogen elimination, results from chelation, which blocks phosphine dissociation and accompanying pathways for b-hydrogen elimination from 14electron, three-coordinate species. Many mechanistic questions remain poorly understood at this point, but these results provide a general, experimentally supported pathway for reactions catalyzed by complexes with monodentate and chelating phosphines.
As described in the section on catalyst development, Xantphos is an e ective ligand for certain aryl halide aminations. This ligand has a particularly large bite angle and a central

1604 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
oxygen that can coordinate to the metal center. Van Leeuwen et al. have recently studied the mechanism of the reaction catalyzed by palladium Xantphos complexes [292]. Several interesting results were found. First, aryl triflate complexes with this ligand are cationic and the central oxygen of the ligand is coordinated to the metal. Second, the reaction of the aryl halide or triflate complexes with amine and base to generate the amido complex is ratelimiting. Oxidative addition is faster than generation of the amido intermediate. These authors deduced that the cationic arylpalladium complexes react by coordination of amine and subsequent deprotonation by the alkoxide base. For reactions proceeding through the neutral arylpalladium bromide complexes with this type of ligand, the mechanism is more complex. Reaction of alkoxide appears to be rate-limiting, and this rate behavior can be attributed to slow reaction of a palladium alkoxide complex with amine to generate the amido species. An accelerating e ect of bromide is observed for this system at low bromide concentrations, and this e ect has been attributed to activation of the alkoxide base by the generation of a less tightly bound ion pair. Alternatively, these authors suggested that higher ionic strength might favor reaction of the arylpalladium halide complex with the alkoxide. At high bromide concentrations, this accelerating e ect was attenuated.
4.7.6.3 Mechanism of Amination Catalyzed by Palladium Complexes with Sterically Hindered Alkyl Monophosphines
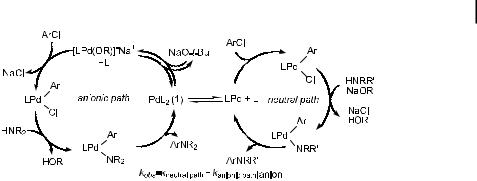
Most recently, Alcazar-Roman and Hartwig have evaluated the amination of aryl chlorides catalyzed by palladium complexes with P(tBu)3 as the ligand [293]. In particular, they were interested in evaluating why couplings of aryl chlorides in the presence of di erent bases occurred at di erent rates when oxidative addition is generally believed to be turnoverlimiting. Their results suggested that the base is intimately involved in the oxidative addition step, most likely by direct coordination of the metal to generate an anionic Pd(0) species. Rate equations were consistent with reaction in the presence of NaOtBu occurring by two mechanisms, one being zero-order- and the other first-order in base.
Amatore and Jutand have shown that reactions of aryl bromides and iodides are accelerated by added halide, and have observed direct coordination of the halide to form an anionic Pd(0) species that undergoes oxidative addition [294, 295]. Thus, the one mechanism that is consistent with the observed first-order behavior in alkoxide and that is consistent
with Amatore and Jutand’s studies involves initial displacement of phosphine by tert-but- oxide to form an anionic Pd(0) species fPd[P(tBu)3](OtBug . Oxidative addition of the aryl chloride would then occur to this anionic species. These concurrent mechanisms are shown
in Scheme 10.
4.8
Summary
The amination of aryl halides and triflates catalyzed by palladium complexes is suitable for use in complex synthetic problems. Many amines couple with a variety of aryl halides and sulfonates to produce high yields of mixed arylamines with at least one of the existing catalyst systems. Nevertheless, there are many combinations of substrates for which the amination chemistry may be substantially improved. For the most part, these reactions involve nitrogen centers, such as those in pyrroles, indoles, amides, pyrazoles, and other heterocyclic groups that are less basic than those in standard alkylamines. Although mild reaction con-
References 161
Scheme 10. Two concurrent mechanisms for the amination of aryl chlorides catalyzed by P(tBu)3 complexes of palladium.
ditions have been developed for many substrates, the harsh conditions used in many of the applications indicate that continued studies on developing milder conditions are warranted. Further, turnover numbers must be improved for the use of this reaction in many industrial applications. In particular, high turnover numbers for reactions conducted with less expensive and weaker bases need to be developed. Finally, mechanistic information is emerging with some of the second-generation catalysts, but data on the most recent catalysts is sparse. Data on reactions of substrates more complicated than simple amines and aryl halides is not presently available.
Acknowledgements
Portions of my groups’ contributions to this work have been supported by the NIH (R29GM55382 and RO1-GM55382) and DOE. We also gratefully acknowledge support from a DuPont Young Professor Award, a Union Carbide Innovative Recognition Award, a National Science Foundation Young Investigator Award, a Dreyfus Foundation New Faculty Award, and a Camille Dreyfus Teacher-Scholar Award for support of this work at its early stages. The author is a fellow of the Alfred P. Sloan Foundation. We also thank Johnson-Matthey Alpha/ Aesar for donations of palladium chloride. Finally, I am deeply indebted to the graduate students, postdoctoral associates, and undergraduates in my laboratory, whose names appear in the references and whose experimental and intellectual input led to our contributions.
References
1 |
R. A. Glennon, J. Med. Chem. 1987, 30, |
5 |
A. G. MacDiarmid, J. C. Chiang, A. F. |
|
1–12. |
|
Richter, A. J. Epstein, Synth. Metals |
2 |
H. Hugel, D. J. Kennaway, Org. Prep. |
|
1987, 18, 285–290. |
|
Proc. Int. 1995, 27, 1–31. |
6 |
A. G. MacDiarmid, A. J. Epstein, Faraday |
3 |
A. Kitani, M. Kaya, J. Yano, K. |
|
Discuss. Chem. Soc. 1989, 88, 317–332. |
|
Yoshikawa, K. Sasaki, Synth. Metals 1987, |
7 |
A. G. MacDiarmid, A. J. Epstein, in |
|
18, 341. |
|
Science and Applications of Conducting |
4 |
F.-L. Lu, F. Wudl, M. Nowak, A. J. |
|
Polymers (Eds.: W. R. Salaneck, D. T. |
|
Heeger, J. Am. Chem. Soc. 1986, 108, |
|
Clark, E. J. Samuelsen), Adam Hilger, |
|
8311–8313. |
|
New York, 1991, pp 117. |

162 4 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
8 |
A. Ray, A. F. Richter, D. L. Kershner, |
28 |
A. J. Paine, J. Am. Chem. Soc. 1987, 109, |
|
A. J. Epstein, Synth. Metals 1989, 29, |
|
1496–1502. |
|
E141–E150. |
29 |
H. Weingarten, J. Org. Chem. 1964, 29, |
9 |
D. Vachon, R. O. Angus, F. L. Lu, M. |
|
977–978. |
|
Nowak, Z. X. Liu, H. Schaffer, F. Wudl, |
30 |
D. M. T. Chan, K. L. Monaco, R.-P. |
|
A. J. Heeger, Synth. Metals 1987, 18, 297– |
|
Wang, M. P. Winters, Tetrahedron Lett. |
|
302. |
|
1998, 39, 2933–2936. |
10 |
M. Stolka, J. F. Yanus, D. M. Pai, J. Phys. |
31 |
P. Y. S. Lam, S. Deudon, E. Hauptman, |
|
Chem. 1984, 88, 4707–4714. |
|
C. G. Clark, Tetrahedron Lett. 2001, 42, |
11 |
E. Ueta, H. Nakano, Y. Shirota, Chem. |
|
2427–2429. |
|
Lett. 1994, 2397. |
32 |
P. Y. S. Lam, G. Vincent, C. G. Clark, S. |
12 |
Y. Kuwabara, H. Ogawa, H. Inada, N. |
|
Deudon, P. K. Jadhav, Tetrahedron Lett. |
|
Noma, Y. Shirota, Adv. Mater. 1994, 6, |
|
2001, 42, 3415–3418. |
|
677–679. |
33 |
S. K. Kang, S. H. Lee, D. Lee, Synlett |
13 |
M. Strukelj, R. H. Jordan, A. |
|
2000, 1022–1024. |
|
Dodabalapur, J. Am. Chem. Soc. 1996, |
34 |
D. J. Cundy, S. A. Forsyth, Tetrahedron |
|
118, 1213. |
|
Lett. 1998, 39, 7979–7982. |
14 |
J. March, Advanced Organic Chemistry; 3rd |
35 |
A. P. Combs, M. Rafalski, J. Comb. Chem. |
|
ed.; John Wiley and Sons, New York, 1985. |
|
2000, 2, 29–32. |
15 |
G. W. Gribble, P. D. Lord, J. Skotnicki, |
36 |
A. P. Combs, S. Saubern, M. Rafalski, |
|
S. E. Dietz, J. T. Eaton, J. L. Johnson, J. |
|
P. Y. S. Lam, Tetrahedron Lett. 1999, 40, |
|
Am. Chem. Soc. 1974, 96, 7812–7814. |
|
1623–1626. |
16 |
P. Marchini, G. Liso, A. Reho, J. Org. |
37 |
P. S. Lam, S. Deudon, K. M. Averill, R. |
|
Chem. 1975, 40, 3453–3456. |
|
Li, M. Y. He, P. DeShong, C. G. Clark, |
17 |
C. F. Lane, Synthesis 1975, 135–146. |
|
J. Am. Chem. Soc. 2000, 122, 7600–7601. |
18 |
H. Heaney, Chem. Rev. 1962, 62, 81–97. |
38 |
W. W. K. R. Mederski, M. Lefort, M. |
19 |
M. Beller, C. Breindl, T. H. Riermeier, |
|
Germann, D. Kux, Tetrahedron 1999, 55, |
|
A. Tillack, J. Org. Chem. 2001, 66, 1403– |
|
12757–12770. |
|
1412. |
39 |
J. P. Collman, M. Zhong, Org. Lett. 2000, |
20 |
R. Rossi, R. H. de Rossi, Aromatic |
|
1233–1236. |
|
Substitution by the SRN 1 Mechanism; |
40 |
J. K. Stille, Angew. Chem. Int. Ed. Engl. |
|
American Chemical Society, Washington |
|
1986, 25, 508–524. |
|
DC, 1983; Vol. 178. |
41 |
J. K. Stille, Pure Appl. Chem. 1985, 57, |
21 |
J. E. Shaw, D. C. Kunerth, S. B. |
|
1771–1780. |
|
Swanson, J. Org. Chem. 1976, 41, 732– |
42 |
A. Suzuki, Pure Appl. Chem. 1994, 66, |
|
733. |
|
213–222. |
22 |
A. J. Pearson, J. G. Park, S. H. Yang, |
43 |
N. Miyaura, A. Suzuki, Chem. Rev. 1995, |
|
Y.-H. Chuang, J. Chem. Soc., Chem. |
|
95, 2457–2483. |
|
Commun. 1989, 1363–1364. |
44 |
E. Negishi, Acc. Chem. Res. 1982, 15, 340– |
23 |
J. P. Collman, L. S. Hegedus, J. R. |
|
348. |
|
Norton, R. G. Finke, Principles and |
45 |
T. Hayashi, Y. Hagihara, Y. Katsuro, M. |
|
Applications of Organotransition Metal |
|
Kumada, Bull. Chem. Soc. Jpn. 1983, 56, |
|
Chemistry, University Science Books, Mill |
|
363–364. |
|
Valley, CA, 1987. |
46 |
T. N. Mitchell, Synthesis 1992, 803–815. |
24 |
A. Klapars, J. C. Antilla, X. Huang, S. L. |
47 |
U. Schopfer, A. Schlapbach, Tetrahedron |
|
Buchwald, J. Am. Chem. Soc. 2001, 123, |
|
2001, 57, 3069–3073. |
|
7727–7729. |
48 |
N. Zheng, J. C. McWilliams, F. J. Fleitz, |
25 |
R. Gujadhur, D. Venkataraman, J. T. |
|
J. D. Armstrong, R. P. Volante, J. Org. |
|
Kintigh, Tetrahedron Lett. 2001, 42, 4791– |
|
Chem. 1998, 63, 9606–9607. |
|
4793. |
49 |
R. Rossi, F. Bellina, L. Mannina, |
26 |
J. Lindley, Tetrahedron 1984, 40, 1433– |
|
Tetrahedron 1997, 53, 1025–1044. |
|
1456. |
50 |
T. Ishiyama, M. Mori, A. Suzuki, N. |
27 |
H. L. Aalten, G. van Koten, D. M. |
|
Miyaura, J. Organomet. Chem. 1996, 525, |
|
Grove, Tetrahedron 1989, 45, 5565–5578. |
|
225–231. |