

Astruc D. - Modern arene chemistry (2002)(en)
.pdf4.6 Applications of the Amination Chemistry 143
ond, Mw values in NMP and THF solutions are dramatically di erent: 24,000–39,000 in NMP and 6300–8200 in THF. Thus, the values in THF would appear to be more realistic. Some incorporation of the BINAP ligand into the polymer product was also observed. A small degree of cross-linking may also be present, although small molecule studies showed only a 0.05 % yield of triarylamine. In addition to the linear poly(m-aniline), a hyperbranched version of this polymer was prepared [227].
Kanbara has since generated a family of poly(iminoarene)s by reaction of 1,3-dibromo- benzene, 4,4-dibromodiphenyl ether, 2,6- and 3,5-dibromopyridines, 2,4-dibromothiophene, and 1,10-dibromoferrocene with a variety of bifunctional arylamines [229]. In many cases, no polymer was obtained, but for polymerizations involving dibromobenzene and 2,6- dibromopyridine, materials with Mn values of over 10,000 were obtained. Spectral data were provided for poly(2,6-aminopyridine) and a polymer made from dibromobenzene and a diarylamino sulfone. These authors have also investigated nickel catalysts for the polymerization of diamines with dichloroarenes, but the materials generated had molecular weights below 10,000 in most cases [230].
Triarylamine polymers that possess oxetane rings for subsequent cross-linking have been generated [231]. N,N-Diphenyl diaminobiphenyl was copolymerized with a dibromo monomer containing the pendant oxetane. Using a catalyst comprised of Pd2(dba)3 and P(tBu)3 at 97 C for 5 d, Braig et al. generated materials with Mn values of 7300–9400 and Tg values of 66–79 C. Films of the polymers were deposited onto indium tin oxide with a photochemically active acid. The materials were then cross-linked under UV irradiation at elevated temperatures and were investigated by UV/vis spectroscopy before and after washing with THF. After heating at 150 C under irradiation, the polymers became insoluble.
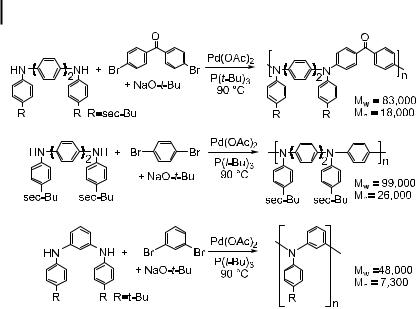
Detailed chemical studies on the formation of triarylamine polymers based on an early communication [232] have now appeared [219]. Goodson, Hauck, and Hartwig reported the use of improved catalysts and synthetic strategies to prepare pure, soluble, linear triarylamine polymers with high molecular weights (Scheme 1). A large number of ligands were prepared and tested for their ability to catalyze the formation of triarylamines in quantitative yields. A phosphino ether ligand and P(tBu)3 were shown to be the most e ective. True polymeric N-aryl versions of poly(p-anilines), poly(m-anilines), alternating poly(m-and p- aniline)s, and alternating donor–acceptor copolymers were all prepared. In the case of polymerizations of substrates with meta-linkages, cyclization to form tetraazacyclophanes occurred in competition with polymerization. The polymerization of oligomeric fragments and/or the separation of the macrocycles from the polymer by size-exclusion chromatography provided purely linear material. Incorporation of phosphine into the polymer by PaC bond cleavage or ligand metalation processes is common in the formation of polymers by palladium-catalyzed cross-coupling [233, 234]. The materials formed with P(tBu)3 as the catalyst contained no phosphorus, as determined by long 31P NMR spectroscopic acquisitions and by 1H NMR spectroscopy. Thus, the use of P(tBu)3 as the ligand is important to observe coupling in yields that are high enough to generate a polymer, to employ low catalyst loads, and to prevent metalation or PaC cleavage reactions of the phosphine ligand.
4.6.2.2 Synthesis of Discrete Oligomers
Buchwald’s and Hartwig’s groups have both used exponential growth strategies to prepare discrete arylamine materials. Buchwald, Sadighi, and Singer prepared oligomers based on
144 4 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
Scheme 1
diarylamine structures [235, 236], while Hartwig and Louie prepared discrete triarylamine oligomers [237]. Janssen has since prepared N-methyl versions of alternating meta,paradiarylamine octamers without exponential growth by coupling dibromobenzene with two tetramers containing amino termini [238]. These materials generate high-spin structures after oxidation. The exponential growth strategy used by Buchwald and Hartwig to generate the larger oligomers relies on orthogonal protection of two functional groups at the termini of the oligomers. Buchwald’s group used N-aryl benzophenone imines as protected anilines and trimethylsilylarenes as masked aryl halides. In addition, Boc groups were used to protect internal diarylamine nitrogens in the monomer units. This protection served to solubilize the materials and to make them less susceptible to air or chemical oxidation. BINAP-ligated palladium complexes were used to conduct the couplings to form the diarylamine linkages. Deprotection of the Boc group and the end-functionalized termini produced materials suitable for studies on the electronic properties of polyanilines with variable chain lengths.
Triarylamine materials of variable chain length [237] were synthesized by using palladium chemistry to couple diarylamines with aryl nonaflates as shown in Scheme 2. The diarylamines were protected with an N-benzyl group, and the aryl nonaflate was masked as a TBSprotected phenol. The TBS-protected phenol was directly converted to the nonaflate by CsF and FSO2C4F9, and the benzyl group was, of course, removed by hydrogenolysis. The building blocks were prepared prior to the use of P(tBu)3 for this chemistry. The CaN bonds of the chain were, therefore, constructed using a complex containing DPPF as ligand; a bromide additive appeared to increase reaction yields, but it is unclear as to how large an e ect this additive had on this amination process. The materials produced were soluble in organic solvents by virtue of the methoxy substituents on the pendant N-aryl groups.
Hyperbranched triarylamine materials have also been prepared by the amination chemistry, as shown in Scheme 3. The material contains exclusively p-phenylene diamine linkages

4.6 Applications of the Amination Chemistry 145
Scheme 2
and triarylamines. The triarylamines were constructed with a combination of benzyl-pro- tected 4,40-dibromodiarylamines and lithium diarylamides. The formation of the triarylamine linkage with aryl bromides and lithium diarylamides proceeded in >90 % yield under mild conditions in the presence of tris(o-tolyl)phosphine-ligated palladium as the catalyst. These materials were characterized by conventional spectroscopic means, as well as by microanalysis. Each carbon was resolved in the 13C NMR spectrum, and a molecular ion was observed by mass spectrometry. The electrochemical behavior of the three-fold symmetric dendrimer was complex and showed a large number of reversible electrochemical waves. The radical cation was generated in solution, which proved to be stable and could be observed by ESR spectroscopy. This material also shows a high glass-transition temperature.
Scheme 3. Synthesis of triarylamine dendrimers.

146 4 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
Multiple arylations of polybromobenzenes have also been conducted to generate electronrich arylamines. Tris(4-bromophenyl)amine and 1,3,5-tribromobenzene both react cleanly with N-aryl piperazines with either P(o-tolyl)3 or BINAP-ligated catalysts to form hexaamine products [130]. Reactions of other polyhalogenated arenes have also been reported [189].
4.6.2.3 Synthesis of Azacyclophanes
In 1998 and 1999, two groups reported macrocyclizations to form triand tetraazacyclophanes [239–242]. These classes of arylamine have more defined structures than the linear oligomers. Tanaka has prepared cyclic, meta-linked trimers of N-methylaniline. Both Tanaka and Hartwig have prepared alternating meta,para,meta,para-tetraazacyclophanes. Tanaka’s structures bear N-methyl groups, while Hartwig’s bear N-aryl groups of varying electronic structure. Both research groups showed that the high-spin state of the dication is the ground state or is energetically low lying relative to the singlet state. The N-methyl cyclophanes generated dications that were unstable except at well below room temperature. The N-aryl cyclophanes were stable for hours at room temperature. The most recently developed catalysts bearing P(tBu)3 as the ligand have led to high yields of the N-aryl tetraazacyclophanes in the last cyclization step and yields of 20–30 % from a one-pot synthesis. The N-methyl materials were produced in less than 5 % yield.
4.6.2.4 Synthesis of Small Molecules for Materials Applications
Palladium-catalyzed amination has also been used to prepare small arylamines that can function as sensors, nonlinear optical materials, magnetic materials, electrode modifiers, hole-transport materials, and dyestu s. N-Arylpolyamines that have the potential for multiple applications have also been prepared.
Azacrown ethers with chromophores bound to the nitrogen for metal cation selective detection systems have been prepared by several groups. Aza-18-crown-6 reacts in modest yields with 9-bromoanthracene in the presence of palladium catalysts to form the N-aryl azacrown [243]. Among reactions conducted with the four ligands P(o-tolyl)3, BINAP, DPPP, and DPPF (DPPP ¼ 1,3-diphenylphosphinopropane), the DPPF as one with ligand furnished the highest yield, 29 %. Improved yields have recently been reported with electronpoor aryl bromides, such as bromobenzonitrile, bromobenzophenone, 2-bromopyridine, and dibromo analogues of the latter two substrates [244]. For these substrates, P(o-tolyl)3 and PPh3 were used as ligands. In addition, Witulski et al. prepared an alkali metal chemosensor by an amination to form a 10-cyano-9-N-anthryl-azacrown ether. The nitrogen donor and cyano acceptor create a material showing intramolecular charge transfer that allows spectroscopic detection of metal ions [245]. Concurrently, Zhang and Buchwald employed dialkyl biphenylylphosphines in the synthesis of N-aryl azacrowns. They showed that ligand 13 generates catalysts with improved activity, relative to those generated from P(o-tolyl)3 or PPh3, for the coupling with azacrown ethers. Beletskaya has used the DPPF-ligated palladium system to conduct selective monoarylation of ethylene diamine, diethylene triamine, triethylene tetraamines, and 2,2-dimethylbutane-1,3-diamine [246]. These polyamines could also be used for ion sensing by attaching chromophoric aryl groups to the polyamine.
A number of groups have used the palladium chemistry to prepare triarylamines that are relevant to electronic materials. Barlow and Marder showed that N,N-diarylaminoferrocenes can be prepared by this methodology. Aminoferrocene [247] was allowed to react with 4- bromotoluene under rigorously anaerobic palladium-catalyzed conditions with DPPF as the
4.6 Applications of the Amination Chemistry 147
ligand to produce N,N-di-p-tolylferrocene in 58 % yield [248]. This material is readily oxidized to the radical cation. The radical resides predominantly on the ferrocene, but the metal–ligand charge-transfer band is red-shifted. Lin, Tao and co-workers have generated green light-emitting carbazole derivatives using the palladium-catalyzed amination [249]. Using catalysts generated from P(tBu)3 and Pd(dba)2 in a 1:1 ratio, they prepared N-phenyl carbazole and, after bromination, a structure with two diarylamino groups on the carbazole. One of the N-aryl substituents of each diarylamino group was a pyrenyl group, while the other was a phenyl, tolyl, or anisyl group.
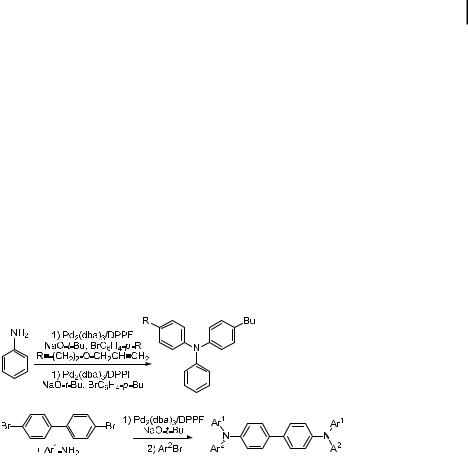
Marder et al. were the first to show that unsymmetrical triarylamines could be prepared, as shown in Eqs. (41) and (42), by sequentially reacting aniline with two di erent aryl halides, or by reacting 4,40-dibromobiphenyl with an arylamine and then a second aryl halide with DPPF-ligated palladium as the catalyst [250]. Buchwald subsequently published similar results on the formation of mixed alkyl diarylamines starting from a primary alkylamine [251] or a primary arylamine [252]. With ligand 15, good yields were obtained in one-pot syntheses of triarylamines. Meerholz used DPPF to prepare TPD analogues containing crosslinkable groups analogous to those used to generate the triarylamine polymers discussed above [253].
ð41Þ
ð42Þ
The amination chemistry has also been applied to dyes. Ipaktschi and Sharifi reported the palladium-catalyzed synthesis of 2,7-diaminofluorenones by two indirect routes due to the base sensitivity of fluorenones [254]. In the first, 2,7-dibromofluorene was initially treated with secondary amines, and subsequent oxidation of the product formed the diaminofluorenone. Alternatively, reaction of aminostannanes derived from secondary amines with 2,7-dibromofluorenone gave yields of the fluorenone ranging from 42–58 %. Most recently, Mullen et al. have prepared thermotropic perylene dyes [255]. Using benzophenone imine as an ammonia surrogate and palladium-BINAP catalysts, they generated an amino-substituted perylene dye from the corresponding brominated dye. After acid-mediated hydrolysis of the benzophenone imine and the introduction of two Boc groups on the nitrogen, the dye changes from one color at low temperature to a second at higher temperature due to release of the Boc groups and regeneration of the free arylamine.
4.6.3
Palladium-Catalyzed Amination in Ligand Synthesis
Amido compounds are gaining increasing importance in early transition metal chemistry as supporting ligands, and structures with both phosphorus and nitrogen donor atoms are

1484 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
finding increased use as non-C2 symmetric ligands for asymmetric catalysis. Thus, the amination chemistry can provide a useful method for ligand synthesis. In the past few years, the number of ligands that have been generated using this chemistry has increased rapidly.
Schrock reported a palladium-catalyzed synthesis of triamido ligands that are useful for performing a-olefin polymerization chemistry with Group IV metals (Eq. (43)) [256]. For Schrock’s group, this reaction has become a mainstay for the synthesis of related ligands for early transition metal chemistry. Arylation and subsequent alkylation of diethylene triamine led to a set of diamido amine ligands with terminal mesityl groups and an internal tertiary amine [257]. In addition, a diamido ether ligand was prepared with N-mesityl groups by the arylation of o,o0-diamino diphenyl ether [258]. The coordination, organometallic, and polymerization chemistry of these Group IV complexes has been studied in depth.
An interesting approach to generating ligands for late transition metal polymerization catalysts has recently been reported by Hicks and Brookhart [259]. A series of 2- anilinotropones was prepared by the amination of a pseudoaromatic triflate. This reaction features a novel substrate for the amination process. As shown in Eq. (44), the uncatalyzed reaction of 2-tosyloxytropone with aniline gave low yields of the desired anilinotropone, but the palladium-catalyzed amination of the corresponding triflate with a variety of anilines gave high yields of the desired material.
ð43Þ
ð44Þ
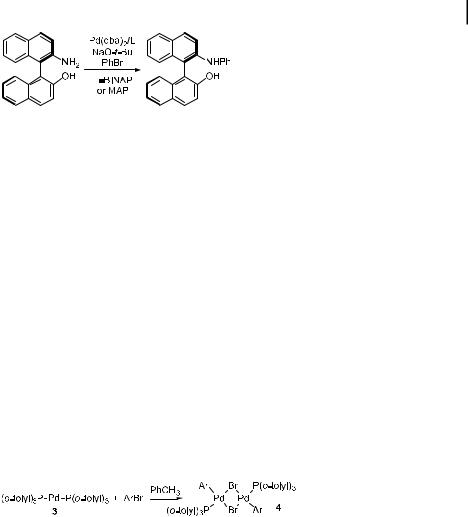
A number of nonracemic, chiral ligands have also been prepared using the aromatic amination process. A small library of C2-symmetric N,N0-diaryldiamine ligands was prepared using BINAP-ligated palladium as the catalyst. A variety of chiral diamines were coupled with a variety of aryl bromides to give the N,N0-diaryldiamines in modest to excellent yields [260–262]. Some of these ligands have been used for Lewis acid-catalyzed reactions [260, 261] and others for transfer hydrogenations [262]. Kocovsky has treated his NOBIN aminoalcohol (Eq. (45)) with bromobenzene to modify this basic ligand structure [155, 263, 264]. Use of the P,N ligand MAP led to significant rate enhancements. Buchwald and Singer have also conducted palladium-catalyzed amination on binaphthyl ligand substructures. They coupled benzophenone imine with the triflate formed from optically pure binaphthol to prepare similar N,O ligands without a need for resolution [265]. Finally, Diver et al. targeted a di erent type of ligand that contained a phenylene backbone and chiral N-alkyl groups [266]. They prepared this ligand by the reaction of dibromobenzene with enantiopure phenethylamine. The highest yields were obtained with ligand 13, and the enantiopurity of the starting phenethylamine was retained in the coupled product.
4.7 Mechanism of Aryl Halide Amination and Etheration 149
ð45Þ
4.7
Mechanism of Aryl Halide Amination
The previous sections described synthetic methods involving palladiumand nickel-catalyzed additions of amines to aryl halides and triflates. The development of procedures and catalysts for these processes has occurred hand in hand with mechanistic analysis of the amination chemistry. The following section describes the current understanding of the mechanistic origin of the e ectiveness of these procedures and catalysts and how this understanding has led to some of the breakthroughs described above.
4.7.1
Oxidative Addition of Aryl Halides to L2Pd Complexes (LFP(o-tolyl)3, BINAP, DPPF) and its Mechanism
L2Pd(0) complexes bearing the three ligands P(o-tolyl)3, BINAP, and DPPF are the resting states of the catalyst when the isolated Pd(0) complex is used as the catalyst in mechanistic studies and when Pd(OAc)2 is used as a catalyst precursor in more synthetically convenient procedures [98, 150]. Pd(0) ligated by dba and BINAP [267] in combination with the L2Pd(0) complex is the resting state of reactions catalyzed by BINAP and Pdn(dba)m complexes [268]. Thus, oxidative addition is the turnover-limiting step with these catalyst systems, and the mechanism and rate of these reactions determines the generation of arylamine products.
ð46Þ
fPd[P(o-tolyl)3]2g undergoes oxidative addition of aryl halides to provide the unusual dimeric aryl halide complexes fPd[P(o-tolyl)3](Ar)(Br)}2 (Eq. (46)) [91, 122]. These complexes remain dimeric in solution, as determined by solution molecular weight measurements, but react as the monomers.
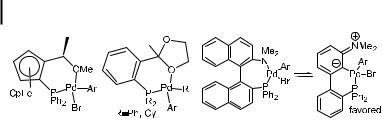
These oxidative addition products have been used as precursors to aryl halide complexes containing several of the P,O and P,N ligands discussed in Section 4.3.3. In these cases, monomeric complexes with or without O- or N-coordination are observed. With these systems, it is di cult to determine whether the heteroatom is coordinated in the complex that lies on the reaction pathway or whether the observed complexes are simply the most stable structures; coordination and dissociation of the heteroatom are generally kinetically undetectable. The three structures obtained are shown in Figure 3. Both Kumada’s [112] and Guram’s [161] ligand systems derived from o-acetyl phosphines generate monomeric, monophosphine structures. Guram’s ligand derived from the o-formyl phosphine forms
150 4 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
Fig. 3. Structures of various aryl halide complexes containing P,O and
P,N ligands used in the amination of aryl halides.
standard monomeric, bis(phosphine) aryl halide complexes. Kocovsky has shown that MAP adopts an unusual conformation involving PdaC coordination [269], which is observed in some binaphthol complexes [270] as part of a complex equilibrium between the expected P,N ligation mode and a third binding mode without C or N coordination.
The mechanism of the oxidative addition of aryl bromides to the bis-P(o-tolyl)3 Pd(0) complex 3 was surprising [271]. It has been well established that aryl halides undergo oxidative addition to L2Pd fragments [272–275]. Thus, one would expect oxidative addition of the aryl halide to occur directly to 3 and ligand dissociation and dimerization to occur subsequently. Instead, the addition of the aryl halide to fPd[P(o-tolyl)3]2g occurs after phosphine dissociation, as shown by an inverse first-order dependence of the reaction rate on the phosphine concentration and the absence of any tris(phosphine) complex in solution [271].
A similar mechanism presumably operates for the oxidative addition of aryl halides to Pd(0) complexes coordinated by two P(tBu)3 ligands, but the thermodynamics of this oxidative addition is unusual. Roy and Hartwig obtained the unexpected result that oxidative addition to Pd[P(tBu)3]2, and presumably to related, highly hindered bis(phosphine) palladium(0) complexes as well, is close to thermonometal or is endoergic [276]. Reaction with aryl chlorides is even less favorable than addition of aryl bromides, which is less favorable than addition of aryl iodides. Although oxidative addition is generally rate-limiting with these systems and the thermodynamics is less favorable for addition to these Pd(0) complexes than for addition to more conventional palladium-phosphine complexes, the rates of addition are su ciently fast to allow room temperature reaction of aryl bromides and chlorides in some cases.
Alcazar-Roman and Hartwig have conducted kinetic studies on the oxidative addition of aryl halides to L2Pd(0) complexes ligated by BINAP and DPPF [268]. It was shown that full dissociation of the chelating ligand occurs to generate an LPd(0) (L ¼ DPPF or BINAP) species prior to oxidative addition of the aryl halide. The relative rates for reassociation of the ligand and addition of aryl halide were depended on the ligand and altered the kinetic behavior of the systems. Thus, the LPd(0) fragment with L ¼ BINAP reacts faster with aryl bromide than with BINAP when the aryl bromide concentration is high. The reactions are zero order in aryl bromide under these conditions. If the (BINAP)2Pd(0) complex is part of the catalytic cycle, this result implies that the catalytic cycle will be zero order in all reagents because the aryl bromide concentration is much higher than the free ligand concentration in synthetic reactions. Under these conditions, ligand dissociation from (BINAP)2Pd is the turnover-limiting step. For complexes of DPPF, ligand reassociation to LPd(0) is faster than aryl bromide addition, making the stoichiometric oxidative addition reversible

4.7 Mechanism of Aryl Halide Amination and Etheration 151
and, therefore, the catalytic cycle first order in aryl bromide and inverse first order in free ligand.
4.7.2
Formation of Amido Intermediates
4.7.2.1 Mechanism of Palladium Amide Formation from Amines
In the original process conducted with tin amides, transmetalation formed the amido intermediate. However, this synthetic method is outdated and the transfer of amides from tin to palladium will not be discussed here. In the tin-free processes, reaction of a palladium aryl halide complex with an amine and a base generates a palladium amide intermediate. One pathway for the generation of the amido complex from an amine and a base would be reaction of the metal complex with the small concentration of amide that is present in the reaction mixtures. However, this pathway seems unlikely considering the two directly observed alternative pathways discussed below and the absence of benzyne and radical nucleophilic aromatic substitution products that would be generated from the reaction of an alkali amide with an aryl halide.
ð47Þ
Paul, Patt, and Hartwig showed that the dimeric aryl halide complexes fPd[P(o- C6H4Me)3](Ar)(Br)g2 react with a variety of amines to form the monometallic, amine-ligated aryl halide complexes fPd[P(o-C6H4Me)3](amine)(Ar)(Br)g in Eq. (47) [122]. Buchwald and Widenhoe er subsequently published similar results and showed that primary amines can even displace the phosphine ligand [192, 277, 278]. It is likely that similar amine-ligated monophosphine complexes are formed when other hindered phosphines are employed, such as the tert-butyl- and cyclohexylphosphine ligands discussed in Section 4.3.3. These amine complexes are important in the catalytic cycle because the acidity of the NaH bond is enhanced upon coordination of the amine to the metal.
ð48Þ
The amine-ligated aryl halide complexes react with alkoxide or silylamide bases to form arylamine products (Eq. (48)) [279]. The reaction of fPd[P(o-C6H4Me)3](HNEt2)(p-Bun)(Br)g and LiN(SiMe3)2 occurred immediately at room temperature to form the arylamine in greater than 90 % yield. Low-temperature reactions conducted in the NMR spectrometer probe allowed direct observation of the anionic halo amido complex fPd[P(o- C6H4Me)3](NEt2)(Ar)(Br)g [279]. Thus, one experimentally supported mechanism for gen-

1524 Palladium-Catalyzed Amination of Aryl Halides and Sulfonates
eration of the amido aryl intermediate is coordination of the amine to form a square-planar 16-electron complex that reacts with the base.
An alternative pathway when soluble alkoxide or silylamido bases are used involves reaction of an arylpalladium halide complex with the alkoxide or silylamide to form an intermediate alkoxide or amide. These complexes can react with amines to form the required amido aryl intermediates. The reaction of fPd(PPh3)(Ph)(m-OH)}2 with primary alkylamines to generate palladium amido complexes and water (Eq. (49)) [70, 280] gave an early indication that the conversion of an alkoxide to an amide could be occurring during the catalytic cycle. These reactions are reversible, but the equilibrium favors the amido complex.
ð49Þ
Alkoxo intermediates may be formed when monophosphines are used, but the stability of the amine complexes favors the mechanism involving deprotonation of the coordinated amine. Instead, the alkoxo complexes are likely to be important in catalytic systems involving chelating ligands [65]. Indeed, the DPPF complex fPd(DPPF)(p-ButC6H4)(OtBu)g reacted with diphenylamine, aniline, and piperidine, as shown in Eq. (50) to give, in each case, the product of amine arylation in high yield [65]. Since no alkali metal is present in this stoichiometric reaction, the palladium amide is formed by a mechanism that cannot involve external deprotonation by an alkali metal base. Of course, reactions involving the carbonate and phosphate bases, which are insoluble and would act as poor ligands, are more likely to occur by a coordination and deprotonation mechanism even when chelating ligands are used.
ð50Þ
4.7.3
Reductive Eliminations of Amines from Pd(II) Amido Complexes
Reductive elimination of amines is the key bond-forming step in the catalytic amination processes. These reactions were unknown a couple of years ago. There are now several examples of this reaction, and the factors that control the rates of this process are beginning to be understood. The identity of the intermediates in some of these reductive elimination reactions has recently been uncovered.