

Emerging Tools for Single-Cell Analysis
.pdfResults |
191 |
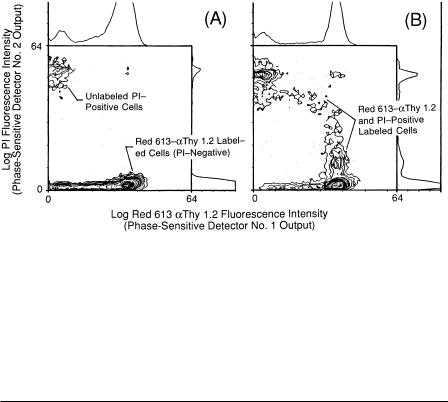
F i g . 8.9. Log phase-resolved fluorescence analysis of Red 613 and PI signals from murine thymus cells labeled with Red 613– Thy 1.2 and PI (“dead cells”) by two PSD channels operating in parallel (A). In
(B), the reference phase shift (φREF1) of PSD channel 1 was first adjusted [φREF1 = /2 φ(PI)] to null signals from thymus control cells stained only with PI and to pass cells labeled with Red 613– Thy 1.2. The
reference phase (φREF2) of PSD channel 2 was next adjusted [φREF2 – /2 φ(Red 613)] to null signals from thymus control cells labeled only with Red 613– Thy 1.2 and to pass cells stained with PI. In (C),
cells labeled with Red 613– Thy 1.2 and stained with PI were analyzed, and the PSDs 1 and 2 signal output histograms were recorded.
were then analyzed at the same reference phase shift and gain settings, and the phaseresolved histograms (see Fig. 8.9C) and corresponding bivariate contour diagram (see Fig. 8.10A) were recorded. Approximately 96.5% of the total thymus cell population was labeled with Red 613– Thy 1.2 and the remaining 3.5% were PI positive. Essentially none of the Red 613–labeled thymus cells were PI positive when analyzed within a half hour after staining (at 4°C on ice) with PI (see Fig. 8.10A). In Figure 8.10B, thymus cells were labeled with Red 613– Thy 1.2 on ice, placed in PBS at 37°C for 6 with PI, and then analyzed. This resulted in a greater portion of Red 613labeled cells initially taking up PI, followed by a loss in Red 613– Thy 1.2 labeling
192 |
Fluorescence Lifetime Flow Cytometry |
F i g . 8.10. Bivariate contour diagrams of murine thymus cells labeled with Red 613– Thy 1.2 and PI (from Fig. 8.9C) (A) and on perturbed cells labeled with Red 613– Thy 1.2 and placed in PBS at 37° with PI (6 h) (B).
intensity of the maximally PI positive cells. These results demonstrate the ability to resolve highly overlapping fluorescence emissions, differing in intensity by greater than 40 times, based on differences in lifetimes.
SUMMARY
Fluorescence lifetime flow cytometry is so new that many of its potential applications have not been fully explored or developed. This technology will add a new dimension to multiparameter flow cytometric analyses through the development of techniques for rapidly measuring fluorescence lifetime of probes bound to macromolecular complexes in cells, increase the range of fluorescent markers that can be used in multilabeling applications, reduce background interferences, and thus enhance measurement precision to yield more accurate results. In the past, procedures were limited in some cases by the availability of fluorescent markers with common excitation regions (so that a single laser excitation source could be used) and emission spectra that were sufficiently separated using optical color-separating filters. Because the lifetime-based sensing technology can separate fluorescence emissions electronically (and also optically), quantify fluorescence lifetimes directly, and make conventional flow cytometric measurements, it has a wide range of technically possible applications. The technology will significantly expand researchers’ understanding of biological processes at the cellular, subcellular, and molecular level, and through clinical and biomedical research, we envision that it will contribute to improving diagnoses, treatment, and understanding the underlying mechanisms of human diseases. In addition, it can readily be added to existing commercial flow cytometry systems, where it can be used for virtually any clinical or research application involving the

References |
193 |
analysis of cells, cell function, or subcellular components through the use of fluorescent markers directed to specific targets.
ACKNOWLEDGMENTS
This work was performed at the Los Alamos National Laboratory, Los Alamos, New Mexico, under the sponsorship of the U.S. Department of Energy, the Los Alamos National Flow Cytometry Resource (National Institutes of Health Grant P41RR013150), and National Institutes of Health Grant R01-RR07855. I thank Nancy M. Lehnert, Carolyn Bell-Prince, and Harry A. Crissman for their assistance in cell preparation and staining.
REFERENCES
Alcala JR, Gratton E, Jameson DM (1985): A multifrequency phase fluorometer using the harmonic content of a mode-locked laser. Anal Instrum 14:225–250.
Aten JA, Kipp JBA, Barendsen GW (1980): Flow cytofluorometric determination of damage to chromosomes from X-irradiated Chinese hamster cells. In Laerum OD, Lindmo T, Thorud E (eds). Flow Cytometry IV. Proceedings of the Fourth International Symposium on Flow Cytometsry. Oslo: Universitetsforlaget, pp 485–491.
Beavis AJ, Pennline KJ (1994): Simultaneous measurement of five-cell surface antigens by five-color immunofluorescence. Cytometry 15:371–376.
Beisker W, Klocke A (1997): Fluorescence lifetime measurement in flow cytometry. Proc SPIE 2982:436–446.
Berndt KW, Gryczynski I, Lakowicz JR (1990): Phase-modulation fluorometry using a frequency doubled pulsed laser diode light source. Rev Sci Instrum 61:1816–1820.
Blair DP, Sydenham PH (1975): Phase sensitive detection as a means to recover signals buried in noise. J Phys E Sci Instrum 8:621–627.
Buican TN, Habbersett RC, Martin JC, Naivar MA, Parson JD, Wilder ME, Jett JH (1991): A new FCM data acquisition and sorting system. Cytometry Suppl. 5:136.
Buican TN (1990): Real-time Fourier transform spectroscopy for fluorescence imaging and flow cytometry. Proc SPIE 1205:126–133.
Castro A, Fairfield FR, Shera EB (1993): Fluorescence detection and size measurement of single DNA molecules. Anal Chem 65:849–852.
Crissman HA, Steinkamp JA (1982): Rapid, one step staining procedures for analysis of cellular DNA and protein by single and dual laser flow cytometry. Cytometry 3:84–90.
Deka C, Steinkamp JA (1996): Time-resolved fluorescence-decay measurement and analysis on single cells by flow cytometry. Appl Opt 35:4481–4489.
Deka C, Lehnert BE, Lehnert NM, Jones GM, Sklar LA, Steinkamp JA (1996): Analysis of fluorescence lifetime and quenching of FITC-conjugated antibodies on cells by phase-sensitive flow cytometry. Cytometry 25:271–279.
Deka C, Cram LA, Habbersett R, Martin JC, Sklar LA, Steinkamp JA (1995): Simultaneous dualfrequency phase-sensitive flow cytometric measurements for rapid identification of heterogeneous fluorescence decays in fluorochrome-labeled cells and particles. Cytometry 21:318–328.
Deka C, Sklar LA, Steinkamp JA (1994): Fluorescence lifetime measurements in a flow cytometer by amplitude demodulation using digital data acquisition technique. Cytometry 17:94–101.
194 |
Fluorescence Lifetime Flow Cytometry |
Demas JN (1983): Excited State Lifetime Measurements. New York: Academic Press.
Durack G, Yu W, Mantulin W, Gratton E (1998): Fluorescence lifetime flow cytometry: A phase-sensitive detection system utilizing a cross correlation technique and digital signal processing. Cytometry Suppl. 9:39.
Feddersen BA, Piston DW, Gratton E (1989): Digital parallel acquisition in frequency domain fluorometry. Rev Sci Instrum 60:2929–2936.
Gadella J, Van Hock A, Visser A (1997): Construction and characterization of a frequency-domain fluorescence lifetime imaging microscopy system. J Fluores 7:35–43.
Gadella TWJ, Jovin TM, Clegg RM (1993): Fluorescence lifetime imaging microscopy (FLIM): Spatial resolution of microstructures on the nanosecond time scale. Biophys Chem 48:221–239.
Glazer AH, Stryer L (1983): Fluorescent tandem phycobiliprotein conjugates. J Biophys 43:383–386.
Gratton E, Jameson DM, Rosato N, Weber G (1984): Multifrequency cross-correlation phase fluorometer using synchrotron radiation. Rev Sci Instrum 55:486–494.
Gratton E, Limkeman M (1983): A continuously variable frequency cross-correlation phase fluorometer with picosecond resolution. Biophys J 44:315–324.
Hiebert RD, Jett JH, Salzman GC (1981): Modular electronics for flow cytometry and sorting: The LACEL system. Cytometry 1:337–341.
Jameson DM, Gratton E, Hall RD (1984): The measurement and analysis of heterogeneous emissions by multifrequency phase and modulation fluorometry. Appl Spectros Rev 20:55–106.
Keating SM, Wensel TG (1991): Nanosecond fluorescence microscopy: Emission kinetics of Fura-2 in single cells. Biophys J 59:1186–202.
Keij JF, Bell-Prince C, Steinkamp JA (1999): Simultaneous analysis of relative DNA and glutathione content in viable cells using phase-sensitive flow cytometry. Cytometry 35:48–54.
Lakowicz JR (1983): Principles of Fluorescence Spectroscopy. New York: Plenum Publishing Corp.
Lakowicz JR, Szmacinski H, Nowaczy KK, Berndt KW, Johnson L (1992): Fluorescence lifetime imaging. Anal Biochem 202:316–330.
Lakowicz JR, Laczko G, Gryczynski I (1986): Two-GHz frequency-domain fluorometer. Rev Sci Instrum 57:2499–2506.
Lakowicz JR, Maliwal BP (1985): Construction and performance of a variable-frequency phase-modula- tion fluorometer. Biophys Chem 21:61–78.
Lakowicz JR, Laczko G, Cherek H, Gratton E, Limkeman M (1984): Analysis of fluorescence decay kinetics from variable-frequency phase shift and modulation data. Biophys J 46:463–477.
Lakowicz JR, Cherek H (1981): Resolution of heterogeneous fluorescence from proteins and aromatic amino acids by phase-sensitive detection of fluorescence. J Biol Chem 256:6348–6353.
Loken MR, Parks DR, Herzenberg LA (1977): Two-color immunofluorescence using a fluorescence activated cell-sorter. J Histochem Cytochem 25:899–907.
Meade ML (1982): Advances in lock-in amplifiers. J Phys E Sci Instrum 15:395–403.
Mitchell G, Swift K (1989): The 48000 MHF, A dual-domain fourier transform fluorescence lifetime spectrofluorometer. Proc SPIE 1204:270–274.
Parson JD, Olivier TL, Habbersett RC, Martin JC, Wilder ME, Jett JH (1993): Characterization of digital signal processing in the DiDAC data acquisition system. Cytometry Suppl. 6:40.
Pinsky BG, Ladasky JJ, Lakowicz JR, Berndt K, Hoffman RA (1993): Phase-resolved fluorescence lifetime measurements for flow cytometry. Cytometry 14:123–135.
Roederer M, De Rosa S, Gerstein R, Anderson M, Bigos M, Stoval R, Nozaki T, Parks D, Herzenberg L, Herzenberg, L (1997): 8 color, 10-parameter flow cytometry to elucidate complex leukocyte heterogeneity. Cytometry 29:328–339.
Sailer BL, Steinkamp JA, Crissman HA (1998a): Flow cytometric fluorescence lifetime analysis of DNAbinding probes. Eur J Histochem 42:19–28.
References |
195 |
Sailer BL, Valdez JG, Steinkamp JA, Crissman HA (1998b): Apoptosis induced with different cycle perturbing agents produces differential changes in the fluorescence lifetime of DNA-bound ethidium bromide. Cytometry 31:208–216.
Sailer BL, Nastasi AJ, Valdez JG, Steinkamp JA, Crissman HA (1997a): Differential effects of deuterium oxide on the fluorescence lifetimes and intensities on dyes with different modes of binding to DNA. J Histochem Cytochem 45:165–175.
Sailer BL, Valdez JG, Steinkamp JA, Darzynkiewicz Z, Crissman HA (1997b): Monitoring uptake of ellipticine and its fluorescence lifetime in relation to the cell cycle by flow cytometry. Exp Cell Res 236:259–267.
Sailer BL, Nastasi AJ, Valdez JG, Steinkamp JA, Crissman HA (1996): Interactions of intercalating fluorochromes with DNA analyzed by conventional and fluorescence lifetime flow cytometry utilizing deuterium oxide. Cytometry 25:164–172.
So PTC, French T, Yu WM, Berland KM, Dong CY, Gratton E (1995): Time-resolved fluorescence microscopy using two-photon excitation. Bioimaging 3:49–63.
So PTC, French T, Gratton (1994): A frequency domain time-resolved microscope using a fast-scan CCD camera. Proc SPIE 2137:83–92.
Spencer RD, Weber G (1969): Measurements of subnanosecond fluorescence lifetimes with a crosscorrelation phase fluorometer. Ann NY Acad Sci 158:361–376.
Steinkamp JA, Keij JF (1999): Fluorescence intensity and lifetime measurement of free and particlebound fluorophore in a sample stream by phase-sensitive flow cytometry. Rev Sci Instrum 70:4682–4688.
Steinkamp JA, Lehnert BE, Lehnert NM (1999a): Discrimination of damaged/dead cells by propidium iodide uptake in immunofluorescently labeled populations analyzed by phase-sensitive flow cytometry. J Immunol Meth 226:59-70.
Steinkamp JA, Lehnert NM, Keij JF, Lehnert BE (1999b): Enhanced immunofluorescence measurement resolution of surface antigens on highly autofluorescent, glutaraldehyde-fixed cells analyzed by phasesensitive flow cytometry. Cytometry 37:275–283.
Steinkamp JA, Parson JD, Keij JF (1998): Progress towards combined phase and amplitude demodulation fluorescence lifetime measurements by flow cytometry. Proc SPIE 3260:236–244.
Steinkamp JA, Lehnert BE, Keij JF (1997): Phase-sensitive detection as a means to recover fluorescence signals from interfering backgrounds in analytical cytology measurements. Proc SPIE 2982: 447–455.
Steinkamp JA, Deka C, Lehnert BE, Crissman HA (1996): Fluorescence lifetime as a new parameter in analytical cytology measurements. Proc. SPIE 2678:221–230.
Steinkamp JA, Crissman HA (1993): Resolution of fluorescence signals from cells labeled with fluorochromes having different lifetimes by phase-sensitive flow cytometry. Cytometry 14:210–216.
Steinkamp JA, Yoshida TM, Martin JC (1993): Flow cytometer for resolving signals from heterogeneous fluorescence emissions and quantifying lifetime in fluorochrome-labeled cells/particles by phasesensitive detection. Rev Sci Instrum 64:3440–3450.
Steinkamp JA, Habbersett RC, Hiebert RD (1991): Improved multilaser/multiparameter flow cytometer for analysis and sorting of cells and particles. Rev Sci Instrum 62:2751–2764.
Steinkamp JA, Habbersett RC, Stewart CC (1987): A modular detector for flow cytometric multicolor fluorescence measurements. Cytometry 8:353–365.
Stöhr M, Vogt-Schaden M (1980): A new dual staining technique for simultaneous flow cytometric DNA analysis of living and dead cells. In: Laerum OD, Lindmo T, Thorud E (Eds). Flow Cytomery, Vol. 4. Bergen, Norway: Universitets-forlaget.
Thomspon RB, Frisoli JK, Lakowicz JR (1992): Phase fluorometry using a continuously modulated laser diode. Anal Chem 64:2075–2078.
Veselova TV, Cherkasov AS, Shirokov VI (1970): Fluorometric method for individual recording of spectra in systems containing two types of luminescent centers. Opt Spectrosc 29:617–618.
196 |
Fluorescence Lifetime Flow Cytometry |
Vigo J, Salmon JM, Viallet P (1987): Quantitative microfluorometry of isolated living cells with pulsed excitation: Development of an effective and relatively inexpensive instrument. Rev Sci Instrum 58:1432–1438.
Vindelov LL, Christensen IJ, Nissen NI (1983): A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry 3:323–327.
Wilkerson CW, Goodwin PM, Ambrose WP, Martin JC, Keller RA (1993): Detection and lifetime measurement of single molecules in flowing sample streams by laser-induced fluorescence. Appl Phys Lett 62:2030–2032.
Zarrin F, Bornhop DJ, Dovichi NJ (1987): Laser doppler velocimetry for particle size determination by light scatter within the sheath flow cuvette. Anal Chem 59:854–860.

Emerging Tools for Single-Cell Analysis: Advances in Optical Measurement Technologies
Edited by Gary Durack, J. Paul Robinson Copyright © 2000 Wiley-Liss, Inc.
ISBNs: 0-471-31575-3 (Hardback); 0-471-22484-7 (Electronic)
9
Application of Fluorescence Lifetime and
Two-Photon Fluorescence Cytometry
Donald J. Weaver, Jr.*, Gary Durack, Edward W. Voss, Jr., and Anu Cherukuri†
University of Illinois at Urbana-Champaign, Urbana, Illinois
INTRODUCTION
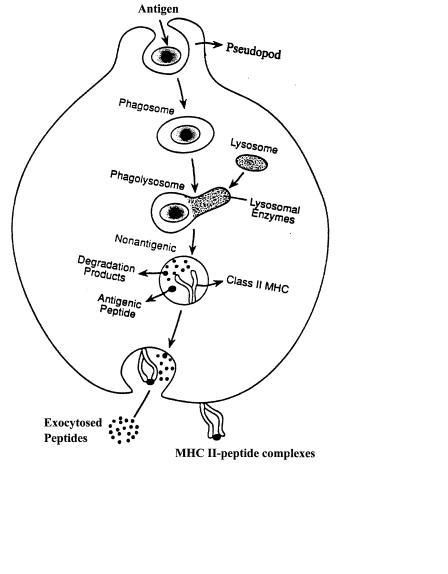
CD4 T-lymphocytes recognize fragments of antigenic proteins in association with major histocompatibility complex (MHC) class II molecules on the surface of cells known as antigen-presenting cells (i.e., macrophages, B cells, and dendritic cells). These antigenic proteins enter the antigen-presenting cell via several processes, including phagocytosis, receptor-mediated endocytosis, and pinocytosis (Lanzavecchia, 1990; Fig. 9.1). Upon internalization, the foreign protein is transported through the endocytic system of the cell (Watts, 1997). As transport occurs, the foreign molecule encounters a variety of biochemical phenomena that serve to denature and degrade the antigen (Germain and Marguiles, 1993; Fig. 9.1). Although alterations in pH and reduction of disulfide bonds facilitate this process, proteolytic cleavage of antigenic proteins into peptides is a fundamental requirement for this pathway (Fineschi and Miller, 1997). Several of the proteolytic enzymes involved in this process have been characterized (Diment, 1993; Fineschi and Miller, 1997). Termed
* Current address: Department of Microbiology and Immunology, University of North Carolina-Chapel Hill, Chapel Hill, North Carolina.
†Current address: Department of Biochemistry, Northwestern University, Evanston, Illinois.
197
198 |
Application of Fluorescence Lifetime and Two-Photon Fluorescence Cytometry |
cathepsins, these enzymes belong to both the cysteine and aspartic families of proteases, and although the precise sequence and site of action of these enzymes have not been clearly elucidated, data suggest that the processing of antigenic proteins by cathepsins is a sequential series of events that results in the formation of 16–21 amino acid fragments capable of interacting with the MHC class II molecule and initiating an immune response to the original protein (Diment, 1993; Fig. 9.1).
As the exogenous protein is degraded, MHC class II molecules are synthesized within the endoplasmic reticulum (ER) of the cell and associate with the nonpolymor-
F i g . 9.1. Classical depiction of the antigen-processing pathway in murine macrophage. Antigenic proteins are internalized into the cell and immediately surrounded by an intracellular vacuole. The antigen-con- taining vacuole proceeds through the endocytic system of the cell. As this occurs, the pH of the vacuole decreases and the disulfide bonds of the protein are reduced. Finally, the protein is degraded into small peptides by the cathepsin family of enzymes. A select few of these peptides are bound by the MHC class II molecule which is also expressed within the endocytic system. Upon peptide binding, the MHC class II-peptide complex is transported to the surface of the cell for recognition by the appropriate T-lymphocyte receptor.

Development of the Problem |
199 |
phic invariant chain as nonameric complexes consisting of three MHC class II heterodimers bound to an invariant chain trimer (Cresswell, 1994; Fig. 9.1). Upon exiting the ER, these complexes are transported to the Golgi and trans-Golgi network for carbohydrate modification prior to entering the cell’s endocytic system (Germain and Marguiles, 1993; Fig. 9.1). Following carbohydrate modification, these complexes are targeted to the endosomal/lysosomal compartments of the cell via a signal sequence in the cytoplasmic tail of the invariant chain and the chain of the MHC class II molecule (Watts, 1997). Intersection of MHC II molecules with the endocytic system provides an opportunity for MHC II molecules to sample a vast array of peptides that have been internalized from various extracellular sources (Watts, 1997). However, the invariant chain must be removed from the MHC class II binding groove prior to peptide binding, and several experiments indicate that this process is dependent upon the same proteases that are responsible for cleavage of antigenic proteins as well as the H-2M chaperonin protein (Cresswell, 1994). Once a stable MHC II–peptide complex is formed, the complex is transported to the plasma membrane for T-cell recognition (Ward and Qadri, 1997; Fig. 9.1). Following stimulation, the activated T cell proceeds to secrete interleukin-2 and upregulate several surface molecules such as CD40 ligand. The ultimate result of these events is the differentiation and expansion of antigenspecific CD4 T-cell clones and subsequent initiation of the immune response.
DEVELOPMENT OF THE PROBLEM
Several questions regarding the intracellular pathways described above remain unanswered. First, little is known about the precise mechanisms that generate antigenic fragments presented by MHC class II molecules (Pieters, 1997). For example, it is unclear if these enzymes act simultaneously or as a sequential cascade (Pieters, 1997). In particular, it is possible that the endopeptidase activity displayed by all cathepsins initiates antigen processing and generates larger fragments containing antigenic epitopes. Subsequently, exopeptidases may be responsible for trimming epitopes to their final size following MHC II binding to ensure preservation of epitopes recognized by T cells. Second, the exact intracellular location for these processing events remains unknown. Numerous research groups have demonstrated that a specialized organelle termed the MHC class II containing compartment (MIIC) is a major component of the processing pathway (Tulp et al., 1994; Qui et al., 1994). However, experiments by Amigorena et al. (1994) demonstrated that a second type of endocytic vesicle termed the CIIV (class II vesicle), which is distinct from the MIIC based on both morphology and marker proteins, is the major compartment for MHC II peptide loading (Amigorena et al., 1994). To reconcile these experiments, several investigators have begun a systematic characterization of the endocytic system of antigen-presenting cells (Kleijmeer et al., 1997). However, since these studies represent a limited number of model antigens and cell types, more rigorous studies are required. Finally, it is imperative that the mechanisms responsible for the transport of these complexes to the surface of the cell be delineated. Initial efforts by Wubbolts et al. (1996) to visualize this process have met with limited success. In short, several

200 |
Application of Fluorescence Lifetime and Two-Photon Fluorescence Cytometry |
issues involved in this pathway remain unresolved. Therefore, a novel experimental approach was required to provide additional insight into this pathway. Since previous experimental strategies relied solely on invasive strategies including electron microscopy and subcellular fractionation, new noninvasive techniques could contribute significantly toward elucidation of this pathway. For this purpose, a novel fluorescent antigenic probe was synthesized and employed.
FLUORESCEIN-DERIVATIZED BOVINE SERUM ALBUMIN AS AN EXOGENOUS ANTIGEN
The probe consists of the small fluorescent hapten fluorescein-5-isothiocyanate (FITC) conjugated to the protein bovine serum albumin (BSA). In the highly derivatized form, fluorescein molecules are relatively nonfluorescent due to their close spatial proximity, orientation, and the short Stoke’s shift between absorption and emission of fluorescein (Fig. 9.2). However, if the spatial constraint is alleviated due to unfolding or proteolytic degradation, a significant increase in fluorescence intensity results (Fig. 9.2). Therefore, FITC-derivatized BSA can be used as a multifunctional probe to monitor several intracellular events, including protein unfolding, disulfide bond reduction, and enzymatic cleavage (Voss et al., 1996). Of major concern in this study was the selection of both the protein carrier and fluorophore. Similar substrates were developed by Rothe et al. (1990) in which synthetic substrates for the enzymes cathepsin L and elastase were conjugated with rhodamine 110 (Rothe et al., 1990; Rothe and Valet, 1993a,b). Bowser and Murphy (1990) conjugated a substrate for cathepsin B with 4-methoxy-β-napthylamine, and upon cleavage, the nonfluorescent substrate was converted to a fluorescent product (Bowser and Murphy, 1990). Although similar in principle, these synthetic substrates had several critical differences from the fluorescein–BSA probe mentioned above. BSA is susceptible to cleavage by a variety of enzymes. Therefore, FITC-derivatized BSA can be used as a proteolytic probe for a vast array of enzymes as opposed to a few specific examples. The antigenic and biochemical properties of BSA are well defined, and BSA typically produces a T-cell-dependent response eliciting high-affinity IgG antibodies (Benjamin et al., 1984). Several studies have also demonstrated that T cells cloned from mice of the H-2d haplotype such as BALB/c were high responders to antigenpresenting cells incubated with BSA (Benjamin et al., 1984). As a result, BSA was an ideal carrier due to its ability to elicit a strong immune response.
The immunogenicity and antigenicity of fluorescein in the context of a carrier protein is well documented (Voss, 1984). Fluorescein typically elicits high-affinity IgG antibodies (Kranz and Voss, 1981; Voss, 1990). In addition, fluorescein possesses several unique and advantageous spectral properties. Specifically, since the fluorescence lifetime of fluorescein is sensitive to alterations in pH, FITC–BSA can be used as a probe to monitor the pH gradient encountered during the endocytic trafficking of proteins with the cell (Fig. 9.3; Ohkuma and Poole, 1978). The fluorescent properties of fluorescein also allow for the measurement of several parameters, including fluorescence polarization, lifetime, and intensity. Therefore, a more complete under-