

Вірусологія
.pdfнезначно різняться своєю будовою, але активні центри в них побудовані однаково.
Рис.6.9. Схема добудови подвійного ланцюга ДНК за допомогою ферменту ДНК-полімерази.
ДНК-полімерази були відкриті в 1955 році Корнбергом зі Станфордського ун-ту, США. ДНК-полімераза виконує ряд функцій, в тому числі забезпечує репарацію та реплікацію ДНК. Ці ферменти здатні подовжувати короткі олігонуклеотидні затравки, приєднуючи до 3’ кінця наступний нуклеотид, але для цього необхідно, щоби затравки були гібридизовані (зв’язані) з комплементарним ланцюгом ДНК, який називається матрицею. Розчин, в якому відбувається ця реакція, повинен містити нуклеозидтрифосфати (вони використовуються як будівельні блоки). Нуклеотид, який приєднує ДНК-полімераза, комплементарний основі у відповідному положенні матричного ланцюга. Наприклад, якщо це А, полімераза приєднує Т, якщо матричний нуклеотид Г, то фермент приєднує Ц. Багаторазово повторюючи цю реакцію, полімераза може подовжувати 3’кінець праймера, поки не дійде 5’ кінця матриці. В ході репарації та реплікації кожен ланцюг виступає матрицею для синтезу іншого.
Для розуміння механізмів ПЛР необхідно відмітити таку властивість ДНК, яка є основою ПЛР – це можливість ДНК денатуруватися (при плавленні) та ренатуруватися (при відпалюванні). Тобто при нагріванні дволанцюнові молекули ДНК розплітаються та утворюються 2 окремі ланцюги, а при охолодженні відбувається їх ренатурація – знову з’єднуються 2 ланцюги за принципом комплементарності (Рис. 6.10).
201
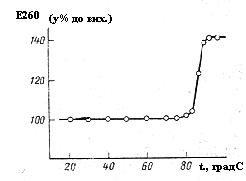
Рис. 6.10. Крива плавлення ДНК бактеріофагу Сд (ДНК бактеріофагу Сд в 0,15М NaCl + 0,015M трицитрат Na (рН 7,0).
При денатурації ДНК in vitro, якщо в суміші присутні олігонуклеотидні мішені (які зазвичай менші за матричну ДНК, тому більш мобільні і скоріше будуть приєднуватися до олДНК) буде відбуватися їх комплементарне з’єднання з матричною ДНК, а ДНК-полімераза миттєво починає синтез (добудову) ланцюга на вільному 3’ кінці.
В 70-роках вчені почали вивчати молекули ДНК за допомогою олігонуклеотидних зондів. Олігонуклеотид – це коротка ділянка нуклеотидів, розташованих в специфічній послідовності. У визначених умовах олігонуклеотид специфічно з’єднується з комплементарною послідовністю нуклеотидів на олДНК. Тому штучно синтезовані радіоактивні олігонуклеотиди можуть слугувати зондами для визначення присутності специфічної послідовності нуклеотидів (радіоізотопний метод). З 1983 року синтез олігонуклеотидів (праймерів) став легким та доступним завдяки автоматичним синтезаторам, які присутні зараз в усіх великих молекулярно-біологічних центрах.
Олігонуклеотидні праймери виконують 2 функції:
З одного боку, вони комплементарні визначеній ділянці ДНК, за іх допомогою можливо знайти специфічну ділянку на молекулі, до якої вони приєднаються за принципом комплементарності, а з другого - вони виступають затравками для початку виконання своїх обов’язків ДНК-полімеразою – добудови ділянки ДНК (що вона успішно робить, використовуючи для цього вільні нуклеотиди).
Зараз найчастіше використовується термостабільна ДНКполімераза, яка попередньо була виділена з бактерії Thermus aguaticus (Tag). Ця бактерія була виділена з гарячих джерел і витримувала температури до 98С. Полімераза, яку
202
використовували в перших експериментах (фрагмент Кленова ДНК-полімерази І E. coli), легко інактивувалася при нагріванні, тому її потрібно було додавати після кожного циклу (раунду реплікації). Полімераза Tag стабільна і активна при високих температурах, тому можливе її додавання один раз на початку ампліфікації. Ще одна перевага Tag полімерази – більш високий оптимум температурного режиму детермінуємої реакції (добудова відбувається при 70-75С), що значно підвищує специфічність, кількісний вихід та можливу довжину синтезованих копій.
Таким чином, до 1983 року були виведені усі посилання для успішного синтезу великої кількості специфічних ділянок ДНК. Для першого досліду з застосуванням ПЛР Кері Міллісом була вибрана ділянка плазмідної ДНК довжиною 25 нуклеотидів та праймери 11 та 13 нуклеотидів. Цей дослід довів, що ампліфікація ділянки ДНК можлива.
Для проведення ПЛР використовують в надлишку олігонуклеотидні праймери, досліджувану ДНК, при цьому утворюється комплекс, та проводять ампліфікацію in vitro. Оскільки праймери гібридизуються з обома ланцюгами ДНК, то і нативна послідовність і синтезовані ПЛР-продукти можуть виступати матрицями для подальших раундів реплікації, в результаті чого число копій унікальної послідовності експоненційно збільшуються (приблизно за формулою 2n , де n число раундів реплікації, які відбулися). Завдяки цьому послідовності, які присутні в досліджуваному матеріалі в мінімальній кількості (одна чи декілька копій) та не виявляються ніякими іншими методами, легко виявляються за допомогою ПЛР. Якщо говорити образно, ПЛР дозволяє знайти "голку в скирді сіна"
– всього одну послідовність ДНК, яка присутня в досліджуваному зразку (Рис. 6.11.).
203
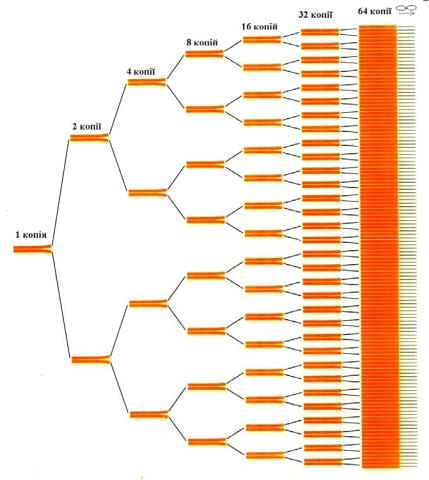
Рис. 6.11. Експоненційне збільшення ділянок ДНК, які утворюються за допомогою ПЛР.
Експоненційне збільшення числа копій послідовності-мішені не тільки забезпечує високу чутливість методу, але й полегшує її виявлення.
Процес ампліфікації складається з повторюючихся циклів температурної денатурації ДНК, відпалу праймерів з комплементарними послідовностями та послідуючою добудовою полінуклеотидних ланцюгів за допомогою ДНК-полімерази. Праймери орієнтовані таким чином, що синтез за допомогою полімерази вібдувається тільки між ними, подвоюючи кількість копій цієї ділянки ДНК (рис. 6.12).
204
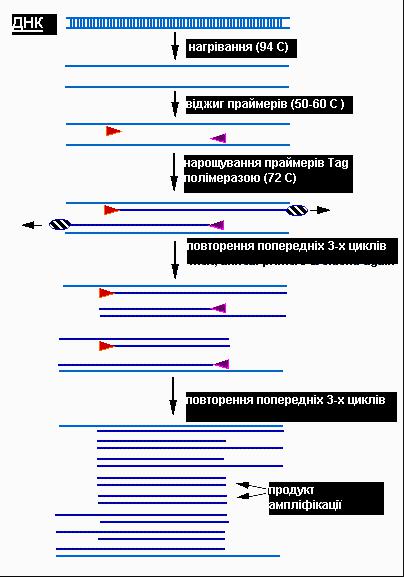
Рис. 6.12. Процес проведення ПЛР (перші декілька циклів ампліфікації).
Після декількох циклів нарощування праймерів, відділення продуктів полімеризації, зв’язування нових праймерів та їх нарощування, довжина ланцюгів ДНК що експоненційно збільшуються (накопичуються) є фіксованою, тому, що їх кінці будуть чітко визначатися 5’кінцями олігонуклеотидних праймерів. Якщо синтезувати праймери, які зв’язуються з більш відділеними одна від одної ділянками, можливо реплікувати молекули більшої довжини.
Обладнання для ПЛР
Реакції зазвичай проводять у 0,2 чи 0,5 мл мікропробірках
205

Епендорф. Обладнання для нагрівання та охолодження може бути зовсім простим, як наприклад набір водяних бань з різною температурою води, якими користувався Керрі Мюлліс, або складним та включати нагрівальний блок, який керується мікропроцесором (Рис. 6.13). Такий блок використовується для автоматичної ампліфікації. Він називається термоциклер, ампліфікатор, або просто ПЛР - машина. Контролюємий комп’ютером блок, розроблений виключно для ПЛР, сьогодні можна придбати у багатьох молекулярно-біологічних фірм.
Рис.6.13 Ампліфікатор на 48 реакцій.
Компоненти реакції.
Для проведення ПЛР необхідні такі складові частини:
•послідовність ДНК, що досліджується;
•буфер;
•дезоксирибонуклеозидтрифосфати (dNTP);
•два праймери, специфічних у відношенні зв’язування
здослідним зразком;
•Tag-полімераза.
Послідовність ДНК, що досліджується, має бути попередньо підготована для аналізу (повинно бути проведено виділення нуклеїнової кислоти з дослідного матеріалу.)
Буферна система має забезпечити оптимальні умови для проведення реакції.
Буфер для ПЛР, при використанні Tag-полімерази , вміщує
50мМ KCl, 10mM Tris-HCl, pH 8,4, 2,5 mM MgCl2 та 100 мкг/мл
206
желатини. При використання комерційних ферментів цей буфер надається разом з ДНК-полімеразою.
Нейтральні розчини dNTP. Краще всього використовувати ліофілізовані порошки і робити з них водні розчини. Обов’язково робити аліквоти в декількох пробірках для можливості часткового використання, зберігати їх при –20С.
Праймери (штучно синтезовані олігонуклеотиди) для ПЛР частіше мають довжину 18-25 нуклеотидів. Можливий синтез і більш довгих затравок, але вони рідко бувають необхідні. Їх можливо синтезувати за допомогою автоматичних синтезаторів ДНК. Кількості олігонуклеотидів, які отримують таким чином (0,2-1 мкмолів) достатні для проведення декількох сотен або тисяч реакцій.
Підбір праймерів – ключова ланка ПЛР, оскільки ними визначається можливість ампліфікації та виявлення необхідної послідовності. ПЛР-ампліфіковані фрагменти ДНК мають різний розмір. Він визначається сумою розмірів праймерів та відстані між їх 3’ кінцями, в більшості випадків
Їх розмір лежить в діапазоні 200-800 нуклеотидів.
Підбір праймерів - процес емпіричний, але відповідність визначеним вимогам збільшує ймовірність отримання працюючих праймерів.
Існують загальні вимоги підбору праймерів:
1.Підбирати праймери необхідно з вмістом ГЦ пар приблизно 50% та випадковим розташуванням основ. (Чим більше ГЦ пар, тим вища температура денатурації).
2.Потрібно уникати послідовностей з стійкими вторинними структурами, оскільки можлива неповна денатурація стійких ділянок. Для вивчення вторинної структури існують спеціальні комп’ютерні програми.
3.Необхідно перевіряти затравки на комплементарність одна одній – очевидно, якщо праймери комплементарно зв’язуються один з одним при відпалюванні, вони не зможуть приймати участь у ампліфікації.
Дизайн праймерів може базуватися як на нуклеотидному, так і на амінокислотному сиквенсі (вироджені праймери).
В залежності від мети експерименту потрібно або відбирати консервативні ділянки (для дослідження вірусів однієї групи), або уникати їх (для дослідження різних штамівнаприклад, якщо праймери розроблені для одного штаму з родини лютеовірусів, то
207
вони не повинні "помічати" інші штами цієї групи, забезпечуючи при цьому ампліфікацію усіх ізолятів вивчаємого).
Для підвищення чутливості та специфічності можливо використовувати так звані "гніздові" праймери (Nested -PCR). При цьому в ролі матриці виступає ділянка ДНК, ампліфікована в першому раунді реплікації, а в другій реакції застосовуються праймери, які ампліфікують ділянку меншу за попередню і розташовану всередині попередньої. Однак такий метод непридатний для рутинної діагностики, він використовується тільки з дослідницькими цілями.
Параметри температурних циклів
ПЛР передбачає інкубацію зразків при трьох температурах, які відповідають трьом етапам циклу ампліфікації – денатурації, відпалу, добудові.
Звичайно длДНК денатурують шляхом короткочасного нагрівання зразку до 90-95С,
потім проводять відпалювання, охолоджуючи зразок до 40-60С (в залежності від довжени праймера та вмісту ГЦ пар). Цю температуру можливо оцінити за формулою:
4˚С х (Г+С) +2˚С х (А+Т)-3).
За рахунок великого надлишку затравок в реакційній суміші гібридизація відбувається майже миттєво та не вимагає довгої інкубації при температурі відпалювання.
Далі суміш нагрівають до 70-75˚С для синтезу (добудови) затравок. Оптимальна температура для роботи Tagполімерази – 72С. Час інкубації при цій температурі залежить від довжини ділянки, що ампліфікується (вважається, що Tag - полімераза синтезує послідовність в 1000 нуклеотидів за 1 хв.)
Аналіз ПЛР-ампліфікованої ДНК
Для аналізу продукту, отриманого в ПЛР, використовують різні методи, такі як: гель-електрофорез, дот- блот-гібридизацію та блот-гібридизацію за Саузерном. З їх допомогою можливо аналізувати більшість ПЛР-продуктів, але абсолютно достовірні результати можливо отримати тільки сіквенуванням.
Частіше всього для швидкої візуалізації результатів ПЛР використовують гель-електрофорез в агарозному гелі за загальноприйнятими методиками.
208
Більшість вірусів рослин, а також віруси тварин є РНК вмісними. Чи можливе вивченя НК таких вірусів та ампліфікація ділянок НК?
Так, можливе, з використанням попередньо отриманої її кДНК копії (за допомогою ферменту зворотньої транскриптази
– РНК-залежної-ДНК-полімерази). Але це набагато складніший процес, оскільки РНК менш стабільна, ніж ДНК. Для діагностики та вивчення РНК-вмісних вірусів користуються методом ЗТ-ПЛТ (RT-PCR). Модифікації можуть бути найрізноманітніші - від попереднього синтезу кДНК з подальшим ампліфікуванням в різних пробірках, до RT-PCR в одній пробірці, що значно спрощує дослідження та підвищує їх чутливість.
Виявлення та детекція віроідів ті вірусоїдів відбувається за допомогою ПЛР, оскільки це – єдина реакція, за допомогою якої можливе швидке та специфічне їх визначення.
Real-time PCR (ПРЛ у реальному часі) та її застосування у вірусологічних дослідженнях.
ПЛР в молекулярній діагностиці широко використовується для детекції нуклеїнової кислоти з будь-яких організмів та займає найважливіше місце в інструментарії дослідницьких лабораторій. Real-time PCR (ПРЛ у реальному часі) походить зі звичайної ПЛР, використовуючи її швидкість, чутливість, повторюваність, при цьому зменшуючи ризик зовнішньої контамінації. В теперішній час використовується 5 хімічних речовин для детекції ПЛРпродуктів протягом real-time PCR. Ці речовини : ДНК зв’язуючі флюорофори, 5’ендонуклеази, суміжні лінійні та булавковидні (hairpin oligoprobes) олігопроби, флюоресціюючі амплікони, які вивчені детально.
Моніторинг акумуляції ампліконів в реальному часі став можливим завдяки міченню праймерів, проб або ампліконів з флюорогенними молекулами. Цей підхід має переваги над радіоізотопними олігопробами, які обов’язково включають радіактивне випромінення.
Швидкість Real Time PCR підвищується за рахунок зменшення часу циклів, відміни пост-ПЛР детекції та використання флуоресцентних міток і чутливих методів детекції
209
їхнього випромінення. Редукція в розмірі ампліконів може також впливати на швидкість реакції, але зменшення розміру продукту не є необхідним для поліпшення точності ПЛР .
Недоліки вживання Real Time PCR в порівнянні з конвенційним ПЛР включають нездатність моніторингу розміру амплікону без входу в систему, несумісність деяких матриць (детекруємих ДНК) з деякими хімічними флюорогенами та високу собівартість аналізу.
Контрольні запитання.
1.З якою метою використовують ПЛР у вірусологічних дослідженнях?
2.Що можливо виявити за допомогою ПЛР?
3.Температурні режими при постановці ПЛР.
4.Основні компоненти полімеразної ланцюгової реакції.
5.Теоретичне обгрунтування можливості постановки ПЛР.
6.Модифікації ПЛР.
7.Виявлення ділянок вірусної РНК за допомогою ПЛР.
8.Чи можливо виявити за допомогою ПЛР пріони?
9.Переваги та недоліки використання ПЛР у реальному часі.
10.Порівняння ПЛР з іншими методами детекції ДНК.
Література.
1.Анализ генома. Методы. Под ред Дейвиса К.- М.:Мир.- 1990.-
С. 176-191.
2.Глик Б., Пастернак Дж. Молекулярная биотехнология. Принципы и применение.- М.: Мир.-2002.-С.181-204.
3.Мюллис К.Необычайная история о том, как родилась полимеразная цепная реакция// В мире науки.- №6.- 1990.-
С.26-34.
4.Molecular methods for virus detection/ Ed. By Danny L. Wiedbrauk.- Academic Press.- 1995.-350p.
5.Molecular biology of plant viruses/ Ed. by C.L.Mandahar.- Kluver Academic Rublisher, USA.-281p.
210