

Matta, Boyd. The quantum theory of atoms in molecules
.pdf72 3 Atomic Response Properties
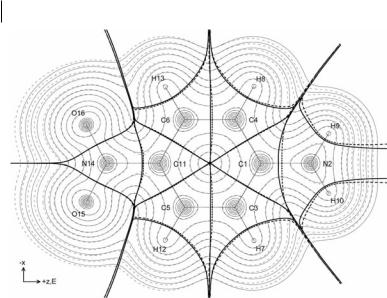
Fig. 3.11 Electron density contours of para-nitroaniline in the nuclear plane, overlaid with interatomic surfaces (bold) and bond paths (semibold).
origin of the molecular coordinate system. For neutral molecules ðQ ¼ 0Þ, m is independent of R0.
As shown in Eq. (1), m can be expressed [2, 3] in terms of atomic dipole polarization contributions, mpðWÞ, and origin-dependent atomic charge-transfer dipole contributions, mcðWÞ:
m ¼ |
Na |
ðW½r R0&rðrÞ dr þ ZW½RW R0& |
|
|
|
X1 |
|
||
|
W |
¼ |
|
|
|
|
|
|
|
¼ |
Na |
ðW½r RW rðrÞ dr þ ½RW R0& ðW rðrÞ dr þ ZW |
||
|
X1 |
|
||
|
W |
¼ |
|
|
|
Na |
ð |
|
|
|
X |
|
||
¼½r RW&rðrÞ dr þ ½RW R0&QðWÞ
W¼ |
1 |
W |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|||
Na |
|
|
|
|
Na |
|
||||
X1 |
fmpðWÞ þ mcðWÞg ¼ |
X1 |
ð14Þ |
|||||||
¼ |
¼ |
|
|
¼ |
mðWÞ ¼ mp þ mc |
|||||
W |
|
|
|
|
W |
|
|
|
||
|
|
|
|
|
|
|
|
|
||
where QðWÞ is the net charge of atom W. Using Eq. (12), the origin-dependent term mcðWÞ can be expressed in terms of bond charges QðWjLÞ as:
NbðWÞ |
|
|
X1 |
ð15Þ |
|
mcðWÞ ¼ ½RW R0&QðWÞ ¼ ½RW R0& |
QðWjLÞ |
|
L¼

3.6 Atomic Contributions to Electric Polarizabilities 73
Using Eq. (8), a corresponding origin-independent expression, mcðWÞ, can be defined, as shown in Eq. (5) and below:
NbðWÞ |
|
X1 |
ð16Þ |
mcðWÞ ¼ ½RW RbðWjLÞ&QðWjLÞ |
L¼
where RbðWjLÞ is the position vector of the bond critical point between atoms W and L.
Note that for each product RbðWjLÞ QðWjLÞ for atom W, there is a corresponding term for atom L which cancels it, because of Eq. (8), and therefore:
Na |
|
Na |
NbðWÞ |
|
|
|
|
|
||||
X1 |
|
X1 |
X1 |
|
|
|
|
|
||||
|
mcðWÞ ¼ |
¼ |
|
|
½RW RbðWjLÞ&QðWjLÞ |
|
||||||
W |
¼ |
|
W |
L |
¼ |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||||
|
|
|
Na |
|
|
Na |
|
|
|
|
||
|
|
|
X1 |
|
|
X1 |
mcðWÞ |
ð17Þ |
||||
|
|
|
¼ |
¼ |
RWQðWÞ ¼ |
¼ |
||||||
|
|
|
W |
|
|
W |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||
|
Na |
|
|
Na |
|
|
|
|
|
|||
|
X1 |
mðWÞ ¼ |
X1 |
|
ðWÞg ¼ mp þ mc |
ð18Þ |
||||||
m ¼ |
¼ |
|
|
fmpðWÞ þ mc |
||||||||
|
W |
|
W |
¼ |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Note that all the quantities in the expressions for mpðWÞ and mcðWÞ are uniquely determined by the molecular charge distribution. An essential criterion for any atomic property is that, once formally defined, its evaluation should be uniquely determined entirely by the molecular wavefunction, just as the evaluation of the corresponding molecular property is.
As an example, Table 3.2 shows the z-axis component of mpðWÞ and mcðWÞ and mðWÞ for each symmetrically unique atom in para-nitroaniline (shown in Fig. 3.11), calculated at HF/6-311þþG(2d,2p)//HF/6-311þþG(2d,2p). The total charge-transfer contribution, mc, is over six times that of the opposing total polarization contribution, mp. The dominant contributor to mc is the NO2 nitrogen, N11, especially the contribution from its bond to the ring, mcðN14jC11Þz.
3.6
Atomic Contributions to Electric Polarizabilities
When a molecule is placed in an electric field, its electron distribution changes in response (nuclear geometry changes are assumed to be negligible here). A useful measure of this response is the molecular dipole moment m, whose first derivative and higher derivatives with respect to the electric field correspond to measurable molecular polarizability and hyperpolarizability tensors [13]. Using QTAIM [1], more detailed information about these molecular responses can be obtained, by partitioning the dipole moment and its electric field derivatives into atomic contributions [3, 4].

743 Atomic Response Properties
Table 3.2 Atomic and bond contributions (in a.u.) to the z-axis dipole moment of para-nitroaniline.
Atom, W |
m(W)z |
mp(W)z |
mc(W)z |
mc(WSL1)zL1 |
mc(WSL1)zL2 |
mc(WSL1)zL3 |
|||
|
|
|
|
|
|
|
|||
C1 |
þ0.381 |
þ0.751 |
0.370 |
0.422 N2 |
þ0.026 C3 |
þ0.026 C4 |
|||
N2 |
0.010 |
þ0.170 |
0.180 |
0.782 C1 |
þ0.301 H9 |
þ0.301 H10 |
|||
C3 |
þ0.101 |
þ0.005 |
þ0.096 |
þ0.024 C1 |
þ0.083 |
C5 |
0.011 |
C6 |
|
C5 |
þ0.144 |
0.030 |
þ0.174 |
þ0.087 C3 |
þ0.128 |
C11 |
0.042 |
H12 |
|
H7 |
0.072 |
0.066 |
0.006 |
0.006 C3 |
|
|
|
|
|
H10 |
þ0.016 |
0.087 |
þ0.103 |
þ0.013 |
N2 |
þ0.126 |
|
þ0.641 |
|
C11 |
þ0.324 |
0.568 |
þ0.892 |
þ0.126 |
C5 |
C7 |
N14 |
||
H12 |
þ0.012 |
þ0.035 |
0.023 |
0.023 |
C5 |
þ0.279 |
|
þ0.279 |
|
N14 |
þ0.927 |
0.922 |
þ1.849 |
þ1.290 |
C11 |
O15 |
O16 |
||
O15 |
þ0.436 |
þ0.146 |
þ0.290 |
þ0.290 N14 |
|
|
|
|
|
Total[a,b] |
þ2.893 |
0.563 |
þ3.456 |
|
|
|
|
|
|
a The analytically calculated value for mðWÞz is þ2.901 a.u.
b An experimental dipole moment for para-nitroaniline (measured in acetone) is 2.44 a.u. [12].
The static dipole polarizability tensor of a molecule, a, is the gradient ð‘EÞ of the molecular electric dipole moment m with respect an external, uniform, and time-independent electric field E, evaluated in the limit of zero field strength:
a ¼ ½‘Em&E¼0 ¼ ½iðqm=qExÞ þ jðqm=qEyÞ þ kðqm=qEzÞ&E¼0 |
ð19Þ |
From Eqs (13) and (19), a is given by: |
|
a ¼ ððr R0ÞrEðrÞ dr |
ð20Þ |
where the electron density derivative rEðrÞ is given by: |
|
rEðrÞ ¼ ½‘ErðrÞ&E¼0 ¼ ½iðqrðrÞ=qExÞ þ jðqrðrÞ=qEyÞ þ kðqrðrÞ=qEzÞ&E¼0 |
ð21Þ |
Throughout this section, the notation tE is used to indicate the gradient of the term t with respect to E, evaluated at E ¼ 0. In terms of atomic contributions we have, from Section 3.5:
Na |
Na |
Na |
|
|||
X1 |
X1 |
X1 |
|
|||
a ¼ |
¼ |
aðWÞ ¼ |
¼ |
mEðWÞ ¼ fmpEðWÞ þ mcEðWÞg |
|
|
W |
W |
W |
¼ |
|
||
|
|
|
|
|||
Na |
|
|
|
|
|
|
X1 |
fapðWÞ þ acðWÞg ¼ ap þ ac |
ð22Þ |
||||
¼ |
|
|||||
W¼

3.6 Atomic Contributions to Electric Polarizabilities 75
The atomic dipole polarization gradient with respect to E, apðWÞ ¼ mpEðWÞ, contains a basin (B) contribution, ap; BðWÞ, arising from the density gradient rEðrÞ within the unperturbed atomic basin. It also contains a surface (S) contribution, ap; SðWÞ, arising from the gradient of the atomic surface S with respect to E:
ð
apðWÞ ¼ ðr RWÞrEðrÞ dr þ ap; SðWÞ ¼ ap; BðWÞ þ ap; SðWÞ ð23Þ
W
Similarly, the gradient of the atomic charge transfer dipole contribution with respect to E, acðWÞ ¼ mcEðWÞ, also contains both basin and surface contributions:
acðWÞ ¼ ac; BðWÞ þ ac; SðWÞ |
ð24Þ |
||
|
NbðWÞ |
|
|
ac; BðWÞ ¼ |
X1 |
ð25Þ |
|
|
f½RW RbðWjLÞ&QBEðWjLÞ |
||
|
L |
¼ |
|
|
|
|
|
|
NbðWÞ |
|
|
ac; SðWÞ ¼ |
X1 |
ð26Þ |
|
|
½RW RbðWjLÞ&QSEðWjLÞ QðWjLÞRbEðWjLÞ |
||
L¼
Using Eqs (7)–(9), the electric field derivatives of the bond charges, QEðWjLÞ, are determined from the electric field derivatives of the atomic charges QEðWÞ:
ð
QEðWÞ ¼ rEðrÞ dr þ QSEðWÞ ¼ QBEðWÞ þ QSEðWÞ ð27Þ
W
The atomic basin and surface bond charge derivative contributions, QBEðWjLÞ and QSEðWjLÞ, can be obtained separately from the corresponding atomic charge derivatives QBEðWÞ and QSEðWÞ.
The contributions to aðWÞ from the surface derivatives with respect to E arise because both the spatial definition of an atom and an atomic dipole moment contribution are determined entirely by the molecular charge distribution, which is of course dependent on E. This is physically sound (it is physically essential), but it also makes the term-by-term evaluation of apðWÞ and acðWÞ more di cult than usual. However, evaluation of apðWÞ and acðWÞ by numerical di erentiation (using finite field wavefunctions) is straightforward. For example:
apðWÞ k ¼ 1=2f½mpðWÞ&E¼ek ½mpðWÞ&E¼0ge 1 |
|
1=2f½mpðWÞ&E¼ ek ½mpðWÞ&E¼0ge 1 |
ð28Þ |

763 Atomic Response Properties
Table 3.3 Atomic contributions (in a.u.) to the principal components of the electric polarizability tensor of para-nitroaniline.
Atom, W |
a(W)zz |
aB(W)zz |
ap(W)zz |
ap, B(W)zz |
ac(W)zz |
ac, B(W)zz |
C1 |
þ9.332 |
þ10.623 |
3.152 |
0.417 |
þ12.484 |
þ11.040 |
N2 |
þ17.854 |
þ16.963 |
0.034 |
þ5.210 |
þ17.888 |
þ11.753 |
C3 |
þ12.865 |
þ13.133 |
2.792 |
þ1.458 |
þ15.657 |
þ11.674 |
C5 |
þ12.077 |
þ12.220 |
4.530 |
þ0.013 |
þ16.607 |
þ12.206 |
H7 |
þ1.885 |
þ1.670 |
þ0.859 |
þ1.164 |
þ1.026 |
þ0.505 |
H10 |
þ1.380 |
þ1.364 |
þ0.848 |
þ0.944 |
þ0.532 |
þ0.370 |
C11 |
þ12.136 |
þ13.844 |
0.580 |
þ1.514 |
þ12.716 |
þ12.330 |
H12 |
þ1.250 |
þ1.004 |
þ0.433 |
þ0.632 |
þ0.817 |
þ0.372 |
N14 |
þ16.944 |
þ15.656 |
3.830 |
0.841 |
þ20.774 |
þ16.497 |
O15 |
þ6.514 |
þ6.310 |
þ4.439 |
þ4.406 |
þ2.074 |
þ1.904 |
Total[a,b] |
þ128.214 |
þ128.487 |
9.083 |
þ22.803 |
þ137.297 |
þ105.685 |
|
|
|
|
|
|
|
Atom, W |
a(W)xx |
aB(W)xx |
ap(W)xx |
ap, B(W)xx |
ac(W)xx |
ac, B(W)xx |
C1 |
þ9.756 |
þ9.830 |
4.362 |
þ0.041 |
þ14.118 |
þ9.789 |
N2 |
þ4.413 |
þ4.435 |
1.590 |
þ1.799 |
þ6.002 |
þ2.637 |
C3 |
þ8.125 |
þ8.385 |
3.283 |
þ1.424 |
þ11.408 |
þ6.961 |
C5 |
þ7.263 |
þ7.768 |
3.220 |
þ1.178 |
þ10.483 |
þ6.591 |
H7 |
þ3.998 |
þ3.558 |
þ0.881 |
þ1.972 |
þ3.117 |
þ1.587 |
H10 |
þ1.258 |
þ1.221 |
þ0.645 |
þ0.778 |
þ0.613 |
þ0.442 |
C11 |
þ9.994 |
þ9.663 |
3.354 |
þ0.637 |
þ13.348 |
þ9.026 |
H12 |
þ3.260 |
þ2.936 |
þ0.845 |
þ1.673 |
þ2.415 |
þ1.263 |
N14 |
þ6.975 |
þ7.367 |
1.430 |
0.494 |
þ8.404 |
þ7.861 |
O15 |
þ7.672 |
þ7.578 |
þ2.925 |
þ3.367 |
þ4.747 |
þ4.211 |
Total[a,b] |
þ94.291 |
þ94.187 |
13.150 |
þ22.766 |
þ107.441 |
þ71.421 |
where the average over numerical derivatives for both positive and negative fields is used. Numerical di erentiation to obtain apðWÞ and acðWÞ, together with the straightforward evaluation of the basin contributions (which involve the analytical density derivatives rE) enables the surface derivative contributions to apðWÞ and acðWÞ to be obtained by di erence. Note that the sum of all of the atomic surface derivative contributions is zero:
Na |
Na |
|
||
X1 |
X1 |
ð29Þ |
||
aS ¼ |
¼ |
aSðWÞ ¼ ½ap; SðWÞ þ ac; SðWÞ& ¼ 0 |
||
W |
W |
¼ |
|
|
|
|
|
||
|
|
|
3.6 Atomic Contributions to Electric Polarizabilities |
77 |
||||
Table 3.3 |
(continued) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
Atom, W |
a(W)yy |
aB(W)yy |
ap(W)yy |
ap, B(W)yy |
ac(W)yy |
ac, B(W)yy |
||
C1 |
þ3.183 |
þ3.291 |
þ3.185 |
þ3.291 |
0.002 |
0.000 |
|
|
N2 |
þ6.950 |
þ6.936 |
þ6.983 |
þ6.936 |
0.033 |
0.000 |
|
|
C3 |
þ6.337 |
þ6.282 |
þ6.340 |
þ6.282 |
0.003 |
0.000 |
|
|
C5 |
þ4.710 |
þ4.796 |
þ4.742 |
þ4.796 |
0.032 |
0.000 |
|
|
H7 |
þ1.115 |
þ1.113 |
þ1.117 |
þ1.113 |
0.002 |
0.000 |
|
|
H10 |
þ0.389 |
þ0.403 |
þ0.375 |
þ0.403 |
þ0.013 |
0.000 |
|
|
C11 |
þ5.136 |
þ4.853 |
þ5.113 |
þ4.853 |
þ0.023 |
0.000 |
|
|
H12 |
þ0.900 |
þ0.904 |
þ0.889 |
þ0.904 |
þ0.011 |
0.000 |
|
|
N14 |
þ2.190 |
þ2.203 |
þ2.140 |
þ2.203 |
þ0.050 |
0.000 |
|
|
O15 |
þ3.497 |
þ3.586 |
þ3.504 |
þ3.586 |
0.007 |
0.000 |
|
|
Total[a,b] |
þ51.354 |
þ51.451 |
þ51.354 |
þ51.451 |
0.000 |
|
|
|
a The analytically calculated values of azz, axx, and ayy are þ128.487, þ94.187, and þ51.451 a.u.
b The experimentally measured isotropic polarizability (measured in acetone) is þ114.73 a.u. [12] compared with the analytically calculated isotropic value of þ91.375 a.u.
Thus, the molecular polarizability a can be written as:
Na |
Na |
|
||
X1 |
X1 |
|
||
a ¼ |
¼ |
aðWÞ ¼ ½apðWÞ þ acðWÞ& |
|
|
W |
W |
¼ |
|
|
|
|
|
||
Na |
|
|
|
|
X1 |
½ap; BðWÞ þ ac; BðWÞ þ ap; SðWÞ þ ac; SðWÞ& ¼ aB þ aS ¼ aB |
ð30Þ |
||
¼ |
|
|||
W¼
That the sum of the atomic surface derivative contributions vanishes does not necessarily mean they should be ignored, however, especially when comparing and/or transferring atomic polarizabilities between molecules.
Using the partitioning method outlined above, Bader et al. [4] studied, among other things, the atomic and group contributions to the polarizability tensors of the normal alkane series and showed that the empirical additivity relationship for the isotropic polarizability of normal alkanes in terms of transferable methylene and methyl group contributions is mirrored by the corresponding theoretical group contributions, thus providing a theoretical basis for the empirical relationship or an experimental validation for the theoretical definition of QTAIM polarizabilities.
As an example here, Table 3.3 shows the diagonal components of apðWÞ and acðWÞ and aðWÞ for each symmetrically unique atom of para-nitroaniline, calculated using the coupled–perturbed HF/6-311þþG(2d,2p)//HF/6-311þþG(2d,2p) level of theory. Also shown in Table 3.3 are the corresponding basin contributions
78 3 Atomic Response Properties
Fig. 3.12 E ect of a finite (0.05 a.u.) electric field applied along the z-axis of para-nitroaniline in terms of electron density contours, interatomic surfaces (bold), and bond paths (semi-bold) in the nuclear plane. Dotted lines correspond to the presence of the field whereas solid lines correspond to the absence of the field.
ap; BðWÞ, ac; BðWÞ, and aBðWÞ. The magnitude of the total charge-transfer contribution to azz is over fourteen times that of the opposing total polarization contribution. The polarizability axx is similarly dominated by charge transfer whereas the much smaller ayy is due entirely to atomic polarization, the charge-transfer contribution vanishing by symmetry (the non-zero values shown for acðWÞyy provide a measure of the error in the results for the other atomic polarizability contributions). That the sum of aðWÞzz does not equal the sum of aBðWÞzz ¼ azz is because of error in the numerical di erentiation used to obtain the total (basin þ surface) aðWÞzz contributions, whereas the aBðWÞzz contributions were calculated from the analytical density derivative. Similar statements apply for the yy and zz components. Figure 3.12 shows the e ect on the charge distribution of a finite external electric field (0.05 a.u.) applied along the z-axis. Note that the shift in electronic charge is opposed to the electric field whereas the shift in the interatomic surfaces is in the opposite direction [3].
3.7
Atomic Contributions to Vibrational Infrared Absorption Intensities
Within the (double) harmonic approximation, the intensity of absorption, I, of a peak in an infrared spectrum for a molecule is proportional to the first derivative

3.7 Atomic Contributions to Vibrational Infrared Absorption Intensities 79
of the electric dipole moment, m, of the molecule with respect to one of the normal-mode vibrational coordinates x (a combination of nuclear displacement coordinates from an equilibrium geometry), in the limit of x ¼ 0. For a normal mode of vibration with frequency nx and normal coordinate x, the corresponding theoretical absorption intensity, Ix, is [13, 14]:
Ix ¼ Cjðdm=dxÞx¼0j2 ¼ Cjmxj2 |
ð31Þ |
where C is a collection of constants and the notation tx is used to indicate the derivative of the term t with respect x, evaluated at x ¼ 0. Absorption intensities of vibrational spectra can be understood in terms of atomic and bond contributions using the equations in Section 3.5 with Eq. (31). The formalism is similar to that for polarizabilities:
Na |
Na |
|
||
X1 |
X1 |
ð32Þ |
||
mx ¼ |
¼ |
mxðWÞ ¼ fmpxðWÞ þ mcxðWÞg ¼ mpx þ mcx |
||
W |
W |
¼ |
|
|
|
|
|
||
mpxðWÞ ¼ ðWðr RWÞrxðrÞ dr þ RWxQðWÞ þ mp; SxðWÞ |
|
|||
|
¼ mp; BxðWÞ þ mp; SxðWÞ |
ð33Þ |
||
NbðWÞ
X
mcxðWÞ ¼ f½RW RbðWjLÞ&Q xðWjLÞ þ RWxQðWjLÞ QðWjLÞRbxðWjLÞg
L¼1
NbðWÞ
X
¼f½RW RbðWjLÞ&QBxðWjLÞ þ RWxQðWjLÞg
L¼1
|
|
|
þ |
NbðWÞ |
|
|
|
|
|
|
X f½RW RbðWjLÞ&QSxðWjLÞ QðWjLÞRbxðWjLÞg |
|
|||
|
|
|
|
L¼1 |
|
|
|
|
¼ mc; BxðWÞ þ mc; SxðWÞ |
ð34Þ |
|||||
|
Na |
|
|
Na |
|
||
X1 |
|
X1 |
ð35Þ |
||||
mSx ¼ |
|
¼ |
mSxðWÞ ¼ |
|
½mp; SxðWÞ þ mc; SxðWÞ& ¼ 0 |
||
W |
|
|
W |
¼ |
|
||
|
|
|
|
|
|||
Na |
|
Na |
|
|
|||
X1 |
|
X1 |
|
||||
mx ¼ |
¼ |
mxðWÞ ¼ |
¼ |
½mpxðWÞ þ mcxðWÞ& |
|
||
W |
|
W |
|
|
|||
|
|
|
|
|
|
||
Na
X
¼½mp; BxðWÞ þ mc; BxðWÞ þ mp; SxðWÞ þ mc; SxðWÞ&
W¼1 |
|
¼ mBx þ mSx ¼ mBx |
ð36Þ |

80 3 Atomic Response Properties
The normal coordinate derivatives of the bond charges QxðWjLÞ and their basin and surface contributions, QBxðWjLÞ and QSxðWjLÞ, are determined from the corresponding atomic charge derivatives QxðWÞ using Eqs (7)–(9). QxðWÞ is given by:
ð
QxðWÞ ¼ rxðrÞ dr þ QSxðWÞ ¼ QBxðWÞ þ QSxðWÞ ð37Þ
W
When compared with the corresponding polarizability term apðWÞ, the term mpxðWÞ contains an additional contribution RWxQðWÞ, which is the atomic charge-weighted derivative of the nuclear position RW with respect to xk. Similarly, the charge-transfer term mcxðWÞ contains an extra contribution for each bond, RWxQðWjLÞ, which is the bond-charge-weighted derivative of the nuclear position RW with respect to coordinate x.
Like the atomic polarizability contributions, the term-by-term evaluation of the atomic polarization and charge-transfer contributions to an electric dipole derivative with respect to a normal vibrational coordinate is complicated (but in a physically necessary way) by the dependence of the atomic surfaces on the nuclear positions. As with the polarizabilities, however, the total mpxðWÞ and mcxðWÞ can each be easily evaluated by numerical di erentiation and the terms involving the analytical density derivative rxðrÞ can be evaluated directly, thus enabling the surface derivative contributions to be obtained by di erence.
Bader et al. [2] were the first to study molecular dipole moment changes associated with normal mode vibrations using QTAIM. Their analysis used finite displacements along a normal coordinate or symmetry coordinate to observe changes in mpðWÞ and an origin-dependent charge-transfer contribution which is somewhat di erent, and less generally applicable, than the mcðWÞ given here, but their results clearly showed that changes in atomic polarizations, changes in atomic charges, and displacement of atomic charges are all important in understanding dipole moment derivatives with respect to normal mode vibrational coordinates, and hence IR spectra.
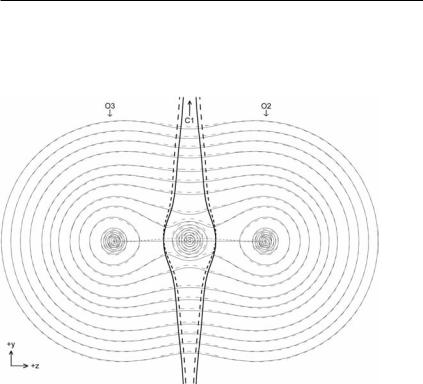
As an example here, Table 3.4 shows the atomic contributions mxðWÞ, mpxðWÞ, and mcxðWÞ to the molecular dipole derivative mx for one of the pair of degenerate bending normal modes of vibration in CO2 and the asymmetric stretching normal mode, calculated at the coupled–perturbed HF/6-311þþG(2d,2p)//HF/6- 311þþG(2d,2p) level of theory. For the bending mode, shown in Fig. 3.13, the calculated dipole derivative is þ0.879 a.u. directed along the y-axis. For the asymmetric stretch, shown in Fig. 3.14, the calculated dipole derivative is 3.793 a.u. directed along the z-axis. The di erence between the calculated dipole derivatives is in good agreement with experimental results [15]. For the bending mode, the atomic polarization derivatives are counter to the larger magnitude atomic charge-transfer derivatives. In contrast, for the asymmetric stretching mode, both the atomic polarization derivatives and the atomic charge transfer derivatives contribute positively to the overall dipole derivative, making it much larger.
|
3.7 |
Atomic Contributions to Vibrational Infrared Absorption Intensities |
81 |
||||||||||
Table 3.4 |
Atomic contributions to the dipole moment derivatives of CO2 |
|
|
|
|
||||||||
|
|
|
|
||||||||||
(in a.u.) with respect to the bend and asymmetric stretch (AS) normal |
|
|
|
|
|
||||||||
mode coordinates. Also shown are the atomic charge Q derivatives.[a,b,c] |
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|||
Atom, W |
mBend(W) |
y |
m |
Bend(W) |
y |
m Bend(W) |
y |
QBend(W) |
|||||
|
|
p |
|
|
c |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
||
C1 |
þ0.191 |
|
0.741 |
|
|
þ0.932 |
|
|
0.012 |
|
|
||
O2 |
þ0.342 |
|
0.795 |
|
|
þ1.137 |
|
|
þ0.006 |
||||
O3 |
þ0.342 |
|
0.795 |
|
|
þ1.137 |
|
|
þ0.006 |
||||
Total |
þ0.876 |
|
2.331 |
|
|
þ3.207 |
|
|
þ0.000 |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Atom, W |
mAS(W) |
|
m |
AS(W) |
z |
|
m |
AS(W) |
z |
|
QAS(W) |
||
|
z |
|
p |
|
|
c |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
||
C1 |
þ1.655 |
|
þ1.213 |
|
|
þ0.442 |
|
|
0.049 |
|
|
||
O2 |
þ1.068 |
|
þ0.780 |
|
|
þ0.288 |
|
|
0.508 |
|
|
||
O3 |
þ1.055 |
|
þ0.898 |
|
|
þ0.157 |
|
|
þ0.573 |
||||
Total |
þ3.778 |
|
þ2.891 |
|
|
þ0.887 |
|
|
þ0.016 |
||||
a Calculated frequencies for the modes are 776 (bend) and 2551 (AS) cm 1.
b Experimental frequencies for the modes are 667 (bend) and 2349 (AS) cm 1. c The experimentally measured ratio of the AS and bend dipole derivatives
is 4.57 [15], compared to the calculated ratio of 4.31.
Fig. 3.13 E ect of finite ðx A0:2 a.u.) bending vibration on CO2 in terms of electron density contours, interatomic surfaces (bold), and bond paths (semi-bold). Dotted lines correspond to bent geometry whereas solid lines correspond to equilibrium geometry.