

Matta, Boyd. The quantum theory of atoms in molecules
.pdf112 4 QTAIM Analysis of Raman Scattering Intensities
Fig. 4.5 QTAIM analysis of the atomic contributions to Da/DrCH in propane for (a) methyl in-plane CH stretch, (b) methyl out-of-plane CH stretch, and (c) methylene CH stretch. Data were calculated at the HF/ D95(d,p) level, from Ref. [17].
ignored. For the stretch of CHip, there is a net increase in the atomic contributions to a from the atoms of the bond being stretched, but a decrease for the remaining atoms. For the CHop and CHm bond stretches, the large positive atomic contributions to Da=DrCH are again from the atoms of the stretched bond, but the Hop and Hm terms are positive for both.
When the atomic contributions are broken down into changes in the CT and AD terms [17], the results are intriguing, although no overriding pattern can be discerned from this small set. For all three bond stretches, the AD term is larger than the CT.
For the CHip stretch, the DAD=DrCH term is positive for both Hip and Cter (terminal carbon, to which it is bonded) but negative for other atoms. The DCT=DrCH term increases for Cter only.
For the CHop stretch, the DAD=DrCH term is large and positive for Hop and Cter but small and negative for the other atoms. The DCT=DrCH term is almost zero for all but the Cter, for which it is only one quarter the magnitude of the value for the CHip stretch.
For the CHm stretch, DAD=DrCH terms are large and positive for Hop and Cm (methylene carbon) and negative for the other atoms. The DCT=DrCH term is again quite small for most atoms. It is larger and positive for Cm and, surprisingly, for Hop.
For methane, ethane and propane, the DAD=DrCH contributions to Da=DrCH are quite similar [17]. As we go from methane to ethane to the CHip stretch in propane, however, the DCT=DrCH of the bonded carbon increases steadily, from 0 in methane, to 0.286 in ethane, to 0.887 in propane. The same term is only

4.6 Patterns in qa/qrCH That Apply Across Di erent Structures, Conformations, Molecular Types 113
0.272 and 0.354 for the bonded carbon in the CHop and CHm stretches, respectively. For the CHip stretch in propane, the xx and yy terms are negligible; almost the entire contribution comes from the zz term: 0, 0.291, and 2.952, respectively. This is intuitively reasonable, because the CHip stretch is almost entirely in the z direction, thus one expects the change in the polarizability to be greatest in this direction.
The trend continues when we compare methane, ethane, and propane with the larger molecules cyclohexane [18] and n-pentane [19]. We begin with the Cter zz contribution to the CHip stretch in pentane. At 1.12, the DAD=DrCH term is larger than for any of the smaller molecules; the most dramatic change, however, is found for the DCT=DrCH term, which is now 10.04. The corresponding terms (xx and yy) for the CHop and CHm stretches are an order of magnitude smaller.
The disparate intensities from CHax and CHeq in cyclohexane set our studies in motion, so it is very satisfying that QTAIM analysis has provided the key that resolves the puzzle. In our arrangement, the CHeq lie in the xy plane and, for the CHeq stretch, the xx þ yy contributions from the C atoms to DCT=DrCH combine to a total of 3.346. This is significantly greater than the corresponding contributions to DCT=DrCH for the CHax stretch. As in propane, this CT term is partly o set by negative contributions at the Heq. The Heq e ectively lie at the end of a pair of short carbon ‘‘chains’’, like the Hip. Although CHax is almost completely aligned with the z-axis, the carbon zz contribution is only 0.420. Despite di erences in the detail, they are more similar to the CHm in propane and pentane.
4.6.1.2 What Did We Learn From QTAIM That Can be Transferred to the Other Molecules?
In parsing the quantity of detail in the preceding sections, we have concluded that it is at least as important to consider the distance across which charge is transferred as to consider the absolute amount of charge transferred. For any CH bond stretch, the change in the molecular polarizability is greatest for the external field that is most closely aligned with the bond. The CHeq in cyclohexane are structurally similar to the CHip bonds in propane and pentane, in that all are aligned with their carbon chains and e ectively lie in the plane of the carbon atoms. Interestingly, these CH bonds are the shortest in the molecule and always produce the greatest Raman scattering intensity. The CHax in cyclohexane have more in common with the CHop and CHm bonds; they are in a plane orthogonal to the carbon framework and are typically slightly longer, with lower isolated CH stretching frequencies and weaker Raman scattering intensities (Fig. 4.1).
From experiment we know that qa=qr (CHip) will be larger than that for any other CH bond in a given molecule, and will increase with chain length. From QTAIM analysis we see that the distinguishing feature is the increase in the DCT contribution of the carbon atom. The molecular polarizability is greatest down the length of the chain; it is also most sensitive to alterations in this dimension. The DAD contributions are mainly confined to the atoms of the bond being

114 4 QTAIM Analysis of Raman Scattering Intensities
stretched. While they too increase with molecular size they do not exhibit, the dramatic di erences found for the DCT(C).
These results form the first of our guiding principles in the Raman scattering intensities – the magnitude of qa=qrCH is highly dependent on bond location within the molecule, orientation relative to the carbon skeleton, and the spatial extent of that skeleton.
4.7
What Can We Deduce From Simple Inspection of qa/qrCH and qa/qrCC From Gaussian?
The larger survey calculations of qa=qr using standard electronic structure calculations include normal and branched alkanes, a diverse selection of cycloalkanes and bicycloalkanes, hedranes and propellanes, and a few alkenes, alkynes, silanes, and extended sheets [22–24]. Some of these are shown in Fig. 4.6. One of our goals was to explore the potential variety of qa=qr values that might exist, seeking extensions to the patterns found above and possible new patterns or factors to consider. A second goal was to identify candidate molecules for which extreme di erences in qa=qr might be observed and which might be amenable to experimental and QTAIM analysis. The CH and CC stretch in all-trans straightchain alkanes up to pentadecane were modeled at the HF/D95(d,p) level [19–21] as well as the Da=DrCH for the gauche-butane, and the gauche–trans and gauche– gauche-pentane conformers. The Da=DrCC were calculated up to n-pentacosane (C25H52). We found that the variety of di erences could be categorized into a few simple patterns that have been discussed thoroughly in our papers. Here we present specific examples to illustrate the factors that were identified.
4.7.1
Variations in qa/qrCH Among the Alkanes
The patterns discovered in the experimental data (Fig. 4.2) are a small part of the larger picture [19, 22, 23].
The stretch of the CHip bond always produces the greatest change in a.
The CHop and CHm bonds, orthogonal to the chain, are all quite similar, though there is a slow, regular decrease from the Da=DrCH in propane with increasing chain length.
Where conformational change rotates the CH bonds to point toward each other, and toward the interior of the molecular skeleton, the value of Da=DrCH decreases. For CH bonds rotated into the plane of the carbon chain Da=DrCH increases (Fig. 4.7).
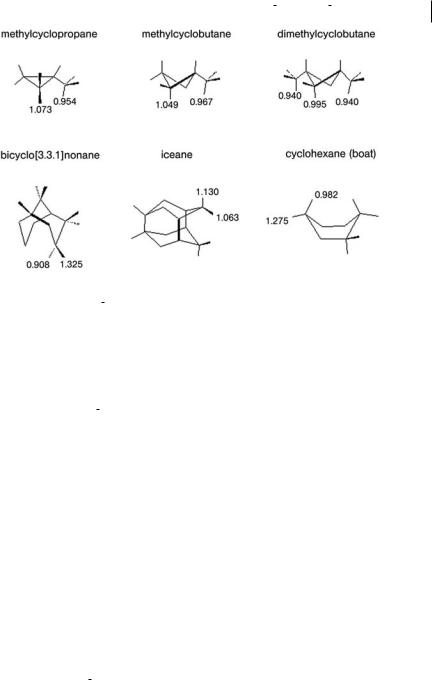
4.7 What Can We Deduce From Simple Inspection of qa/qrCH and qa/qrCC from Gaussian? 115
Fig. 4.6 E ect of conformation and structure on the magnitude of qa/qrCH. Exo/endo di erences are illustrated in the boat form of cyclohexane, steric hindrance in methylcyclopropane, methylcyclobutane, and 1,3-dimethylcyclobutane, and the combined
e ects of axial/equatorial and exo/endo are evident in some of the CH bonds of bicyclo- [3.3.1]-nonane and tetracyclo-[5.3.3.12; 6.04; 9]- dodecane, also called iceane. Data were calculated at the HF/D95(d,p) level, from Refs. [20, 21].
At 1.118, Da=DrCH for the methine CH bond is largest in isobutane. It has the ‘‘in-plane’’ alignment; the methyl CH bonds are in-plane (1.107) or out-of-plane (1.000) (Figure not shown).
QTAIM analyses have not been performed on most of these molecules. We speculate that the pattern of end-to-end CT, opposed by increased ADs at the carbons atoms will be the overriding factor. There is a gradual leveling with chain
Fig. 4.7 E ect of molecular conformation on the magnitude of Da/DrCH in gauche n-butane and gauche-trans n-pentane. The steric hindrance imposed by internal rotation reduces the derivative from approximately 1:05 10 30 Cm V 1 for the methyl out-of-
plane and methylene CH. Rotation of a CH into an end-of-chain in-plane position raises the derivative to a value close to those of the Hip in all-trans butane and pentane (1.207 and 1.26, respectively). Data were calculated at the HF/D95(d,p) level, from Refs. [19, 20].

116 4 QTAIM Analysis of Raman Scattering Intensities
length, as a result of either the damping e ect of intervening methylenes or the unfavorable nature of large CT over greater distances.
4.7.2
Da/DrCH in Cycloalkanes, Bicycloalkanes, and Hedranes
Alkane rings contain additional categories of CH bond, simplified as axial, equatorial, exo, endo and bridgehead. The CHax and CHeq have already been addressed in cyclohexane. The points below represent the cycloalkane variations on the trends discovered in the straight chains (Fig. 4.6).
The di erence between CHax and CHeq increases with ring size.
Steric crowding and rotation such that bonds are directed toward the interior of the skeleton results in a decrease in Da=Dr, whether the bonds are part of the ring or on methyl substituents.
Exo/endo di erences appear, as seen in the boat form of cyclohexane.
Individual e ects identified in simple structures combine in more complicated molecules.
Bicyclo-[3.3.1]-nonane and tetracyclo-[5.3.1.12; 6.04; 9]-dodecane (iceane) serve to illustrate the combined e ects of axial, equatorial, exo, and endo characteristics. The most dramatic di erence in qa=qr was predicted and experimentally verified in bicyclo-[1.1.1]-pentane [24]. The bridgehead CH experiences the combined enhancement of the terminal position and the strain at the carbon atom, to give the largest qa=qrCH of any molecule in this category, irrespective of size.
4.7.3
Patterns That Emerge in Da/DrCC of Alkanes
The unique CC bonds in straight-chain alkanes and in simple cycloalkanes may be easily examined with HF and DFT methods [21–23]. On stretching the skeleton CC bonds we observe complete reversal of the patterns seen in the CH bonds. The magnitude of Da=DrCC is smallest when bonds at the outer ends of the chain are stretched, becoming larger as we progress towards the center of the chain. Even more curiously, the smallest derivative occurs at the second CC bond from the end, as if the terminal position imparts a slightly greater looseness to the charge density. This is not an artifact of the lower level basis set, because the calculations were repeated with B3LYP/aug-cc-pVDZ and identical patterns appeared (Fig. 4.8). With QTAIM analysis, we hope to gain insight that will explain these predictions.
For the ring and cage molecules Da=DrCC were calculated for those bonds for which structural and geometric independence could be maintained. In general, the derivatives for CC bonds in the rings were smaller than for any of the open

4.7 What Can We Deduce From Simple Inspection of qa/qrCH and qa/qrCC from Gaussian? 117
Fig. 4.8 Patterns in Da/DrCC in the straight-chain alkanes. Derivatives for each CC bond, numbered from the beginning of a chain (C1–
C2 ¼ bond 1). Data were calculated at the HF/D95(d,p) level for propane (C3), pentane (C5), nonane (C9), pentadecane (C15), and pentacosane (C25). Data taken from Ref. [21].
chains, from a low of 0.904 for the vertical bonds in hexaprismane to a high of 1.293 for the ring CC bonds in 1,4-dimethylcyclobutane. An increase in the calculated strain energies correlated well (r2 ¼ 0:58, P < 0:0001 for n ¼ 21) with the decreasing Da=DrCC [21], but this is clearly only part of the story for these complicated structures.
4.7.4
Unsaturated Hydrocarbons and the Silanes: CxH, CyC, and SixSi Derivatives
In keeping with the electrically conductive nature of polyenes and silanes, we observe much larger Da=Dr for these molecules [22]. The Da=DrCH for the alkenes follow the same overall patterns as for their saturated counterparts, in that the stretch of the terminal CHip bond produces the largest change in a, but the magnitude falls o much more rapidly for the CH bonds along the chain. The Da=DrCbC values are very large and increase rapidly with chain length (Fig. 4.9).
Fig. 4.9 Patterns in Da/DrCH and Da/DrCC in trans-1,3-butadiene and trans,trans-1,3,5-hexatriene. The end-of-chain e ect is magnified by the polarizability of the p orbitals down the length of the chain, and reduced perpendicular to the chain, compared with similar values for the straight-chain alkanes. Data taken from Ref. [22].

118 4 QTAIM Analysis of Raman Scattering Intensities
Our silane calculations include only up to Si5H12 (data not shown [22]). For Da=DrSiH and Da=DrSiaSi the pattern is the same as for the alkanes – the derivative is largest for SiHip and increases with chain length. The derivatives for the SiHop bonds are similar to those along the chain and decrease with chain length.
4.8 Conclusion
Results from QTAIM analysis for the molecular polarizability derivative performed on a catalog of molecules can be summarized in two principal conclusions:
1.The elements of structure, viz. alignment (cis/trans, eq/ax, in-plane/out-of-plane), steric hindrance, and strain, are local in nature. For rings, strain applies to every atom of that ring.
2.The e ects of these local features are distributed across the molecule.
In a way, QTAIM analysis of qa=qr only serves to emphasize that the molecular wavefunction is a function of all nuclear and electronic degrees of freedom simultaneously.
The QTAIM atoms are as physical as the molecules from which they are recovered, arising naturally from the principles of quantum mechanics. Chemical intuition, based on experiment and theory, relies on identification of the atomic constituents of molecules as a first step, and then on application of established patterns of chemical connectivity, or bonding, to obtain principles of molecular function from structure. We, as chemists, do not work in Hilbert space, but in real space. Our building blocks are not state vectors but concrete, physical atoms. QTAIM coupled with quantum chemical calculations serves as a test bench to explore the intricacies of structure and the interplay between that structure and the resulting molecular properties. When interesting properties appear, or fail to appear, from novel structures, experiment can follow and be supported by a sound theoretical foundation. When interesting properties appear in seemingly standard structures devoid of exotic structural motifs, the need for theoretical analysis is equally clear.
Measurement of absolute Raman scattering intensities and their accurate reproduction through QTAIM are both challenging tasks. QTAIM informs our existing notions of conformation with an understanding based on the atom-by-atom contributions to the changes in molecular properties that arise from local environmental e ects. It turns out for qa=qr that the molecular framework itself is imprinted on the molecular response. Knowledge of the underlying atomic-level topological features of the electron density gives insight into how to design desired function into molecular systems. We are free to modify the local environments of functional groups in any way we choose, including the doping of crys-

References 119
tals. The operating principle is that a local environmental perturbation has a global e ect on a molecular property.
We began this chapter with an investigation of anomalous Raman scattering intensities from CH functional groups. The first impression might have been that this was a rather esoteric probe of a mundane group of atoms. Through our QTAIM analysis we have seen that the Raman intensity is a sensitive probe of structure-imprinted, structure-mediated charge flow and rearrangement within the constraints imposed on the molecular framework by the topology of the electron density.
Authors’ Note
It has become common in the literature to see the QTAIM atoms referred to as ‘‘Bader’s atoms’’. It is a testament to his belief in the supremacy of experimental observation and the rigors of quantum mechanics that Bader himself decries this tendency. They are not some construct arising out of an artificial and arbitrary partitioning of space; they are the quantum mechanical atoms.
References
1R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University
Press, New York, 1990.
2 T. S. Koritsanszky, P. Coppens, Chem.
Rev. 2001, 101, 1583–1627.
3G. Herzberg, Molecular Spectra and Molecular Structure II. Infrared and Raman Spectra of Polyatomic Molecules, Van Nostrand Reinhold Co.
Inc., New York, 1945.
4 E. B. Wilson, J. C. Decius, P. C.
Cross, Molecular Vibrations, McGraw–
Hill, New York, 1955.
5G. Fogarasi and P. Pulay (Ed.: J. R. Durig), Vibrational Spectra and
Structure, Elsevier, 1985, 14, Chapter 3.
6 K. M. Gough, W. F. Murphy, J. Chem. Phys. 1987, 87, 1509–1519.
7D. A. Long, Proc. Roy. Soc. London, Ser. A. 1952, 217, 203; M. Gussoni (Ed.: R. J. H. Clark, R. E. Hester),
Advances in Infrared and Raman Spectroscopy, Heyden & Son, London,
1980, Vol. 6.
8K. M. Gough, W. F. Murphy, J. Chem. Phys. 1986, 85, 4290–4296.
9K. M. Gough, W. F. Murphy, T. Stroyer-Hansen, E. Norby-Svendsen,
J. Chem. Phys. 1987, 87, 3341–3345.
10 W. F. Murphy, J. M. FernandezSanchez, K. Raghavachari, J. Phys. Chem. 1991, 95, 1124–1139.
11 K. M. Gough, H. K. Srivastava, J. Phys. Chem. 1996, 100, 5210–5216.
12 K. M. Gough, W. F. Murphy, J. Mol. Struct. 1990, 224, 73–88.
13 R. Dawes, K. M. Gough, J. Chem. Phys. 2004, 121, 1278–1284.
14 R. L. McCreery (Ed.: J. D. Winefordner), Raman Spectroscopy for Chemical Analysis, Vol. 157 in Chemical Analysis, Wiley Interscience, 2000.
15 G. Placzek; U. S. Atomic Energy Commission, UCRL-Trans-524(L); 1962. Translated from Handbuch der Radiologie, 2nd edn, Marx, E. Ed.; Akademisch: Leipzig, 1934, Vol. 6, Part II, 205–374.
16 R. G. Snyder, A. J. Aljibury, H. L. Strauss, H. L. Casal, K. M. Gough, W. F. Murphy, J. Chem. Phys. 1984, 81, 5352–5361.
120 |
4 QTAIM Analysis of Raman Scattering Intensities |
|
|
|
|
|
K. M. Gough, H. K. Srivastava, K. |
|
M. J. Frisch et al., Gaussian 03, |
17 |
25 |
|||
|
|
Belohorcova´, J. Chem. Phys. 1993, 98, |
|
Revision A.1, Gaussian, Inc., |
|
|
9669–9677. |
|
Pittsburgh PA, 2003. |
18 |
K. M. Gough, H. K. Srivastava, K. |
26 |
F. Biegler-Ko¨nig, AIM2000, |
|
|
|
Belohorcova´, J. Phys. Chem. 1994, 94, |
|
University of Applied Sciences, |
|
|
771–776. |
|
Bielefeld, Germany. |
19 |
K. M. Gough, H. K. Srivastava, J. Phys. |
27 |
K. M. Gough, J. Chem. Phys. 1989, 91, |
|
|
|
Chem. 1996, 100, 5210–5216. |
|
2424–2432. |
20 |
K. M. Gough, J. R. Dwyer, J. Phys. |
28 |
R. Dawes, Ph.D. Thesis, University of |
|
|
|
Chem. A. 1998, 102, 2723–2731. |
|
Manitoba, 2004. |
21 |
K. M. Gough, J. R. Dwyer, R. Dawes, |
29 |
M. G. Papadopoulos, J. Waite, A. D. |
|
|
|
Can. J. Chem. 2000, 78, 1035–1043. |
|
Buckingham, J. Chem. Phys. 1995, |
22 |
C. Lupinetti, K. M. Gough, J. Raman |
|
102, 371. |
|
|
|
Spectroscopy. 2002, 33, 147–154. |
30 |
Y. Le Du , W. Holzer, J. Chem. Phys. |
23 |
K. M. Gough, C. Lupinetti, R. Dawes, |
|
1974, 60, 2175–2178. |
|
|
|
J. Comput. Meth. Sci. Eng. 2004, 4, |
31 |
S. Montero, D. Bermejo, Mol. Phys. |
|
|
597–609. |
|
1976, 32, 1229. |
24 |
R. Dawes, K. M. Gough, J. Chem. |
32 |
K. Kerl, H. Hausler, Ber. Bunsenges. |
|
|
|
Phys. 2004, 121, 1278–1284. |
|
Phys. Chem. 1984, 88, 992. |

121
5
Topological Atom–Atom Partitioning of Molecular Exchange Energy and its Multipolar Convergence
Michel Rafat and Paul L. A. Popelier
5.1 Introduction
Without force fields the structure and dynamics of sizeable systems (biopolymers or condensed matter) would be beyond the predictive power of present day computers. The development of force fields has a long and successful history, culminating into products such as AMBER [1], CHARMm [2] or GROMOS [3], to name just a few. Despite their ubiquitous use and success they define a paradigm of accuracy, largely accepted by a community of users, despite known inherent limitations. It is very di cult to alter this paradigm and achieve a new level of accuracy unless the underlying principles of force field design are also altered, possibly drastically. That there is an increasing urgent need for sustained research e ort in this direction is backed up by a recent review [4] of Ponder and Case on force fields for protein simulations. They concluded that ‘‘An increase in computer power of at least two orders of magnitude should occur over the next decade. Without further research into the accuracy of force-field potentials, future macromolecular modeling may well be limited more by validity of the energy functions, particularly electrostatic terms, than by technical ability to perform the computations.’’
There are several keys to a much needed alternative approach, all based on sound principles. The first key is replacing the intrinsically limited point charges by multipole moments. The second key is to sample as much information as possible from reduced density matrices (both 1st and 2nd order, starting with the electron density). This means that molecules and molecular complexes provide the information with which the force field is endowed, fitted, or trained. This line of attack come under the supermolecular rather than perturbation approach. The third key is a robust partitioning method to generate atomic information. Quantum chemical topology (QCT) [5, 6] is chosen, given its deep roots in quantum mechanics, its widespread use, and because it generates finite nonoverlapping atoms. A fourth key, which we mention in passing, is the use of neural networks to capture the fluctuation of multipole moments in response to