

Matta, Boyd. The quantum theory of atoms in molecules
.pdf162 6 The ELF Topological Analysis Contribution to Conceptual Chemistry and Phenomenological Models
108 |
M. E. Alikhani, F. Fuster, B. Silvi, |
113 |
R. F. W. Bader, R. J. Gillespie, P. J. |
|
Structural Chemistry 2005, 16, |
|
MacDougall, J. Am. Chem. Soc. 1988, |
|
203–210. |
|
110, 7329–7336. |
109 |
B. Silvi, J. Mol. Struct. 2002, 614, |
114 |
R. J. Gillespie, I. Bytheway, T.-H. |
|
3–10. |
|
Tang, R. F. W. Bader, Inorg. Chem. |
110 |
K. S. Pitzer, J. Am. Chem. Soc. 1946, |
|
1996, 35, 3954–3963. |
|
67, 1126–1132. |
115 |
A. L. Allred, E. G. Rochow, J. Inorg. |
111 |
P. G. Mezey, Can. J. Chem. 1993, 72, |
|
Nucl. Chem. 1958, 5, 264. |
|
928–935. |
116 |
S. Shaik, P. Maitre, G. Sini, P. C. |
112 |
S. Diner, P. Claverie, in Localization |
|
Hiberty, J. Am. Chem. Soc. 1992, 114, |
|
and Delocalization in Quantum |
|
7861–7866. |
|
Chemistry (Eds. O. Chalvet, R. Daudel, |
117 |
R. J. Gillespie, E. A. Robinson, Inorg. |
|
S. Diner, J. P. Malrieu), Reidel, |
|
Chem. 2004, 43, 2318. |
|
Dordrecht, vol. II, 1976 pp. 395– |
118 |
D. B. Chesnut, J. Phys. Chem. A 2000, |
|
448. |
|
104, 11644–11650. |
Part II
Solid State and Surfaces

165
7
Solid State Applications of QTAIM and the Source Function – Molecular Crystals, Surfaces, Host–Guest Systems and Molecular Complexes
Carlo Gatti
7.1 Introduction
This chapter deals with the application of QTAIM to the solid state, except for the last section, in which the source function – a recently developed tool for the QTAIM study of the chemical bond from a somewhat original viewpoint – is introduced.
The chapter starts with an illustration of TOPOND software, which implements QTAIM for systems periodic in 0 to 3 dimensions, which covers polymers, surfaces, and crystals, besides molecules. The interface of TOPOND to the multipolar package XD is also mentioned, because it enables QTAIM analysis of experimentally derived electron densities. The chapter continues with an example of a didactic application of TOPOND to a study of crystal field e ects on bonding and on molecular properties in the urea molecular crystal. Clean and chemisorbed semiconductor surfaces then serve as an example of the wealth of information provided by QTAIM about the e ect of surface formation and reconstruction on the bonding and atomic properties of first surface layers. Guest–host systems are discussed as a last example, with emphasis on guest to/from host electron transfer and on the peculiar features of guest–host bonding interactions. The relevance of these key issues to materials science applications is briefly touched upon for thermoelectric materials.
In the last section, the source function (SF) is introduced and examples of its preliminary and future potential applications and extensions are presented. This function enables the value of the density at any point within a system to be equated to a sum of atomic contributions, thus enabling the properties of the density to be viewed from a totally new perspective. Although depending on the whole set of interactions within a system, a bond path is topologically associated only with the two atoms it connects. In contrast, the source function details how all the other atoms in a system, in addition to the two linked atoms, contribute to the accumulation of electron density along a bond path and, in particular, to BCP. It thus discloses nonlocal information on bonding and on complex bonding pat-

166 7 Solid State Applications of QTAIM and the Source Function
terns, analogously to the QTAIM delocalization index or the synaptic order of an ELF valence basin. One advantage of the SF over these two powerful interpretive tools is that it is directly amenable to experimental determination, since to evaluate it only the knowledge of the system’s electron density and Laplacian is required.
7.2
QTAIM Applied to Solids – the TOPOND Package
TOPOND [1–3] has some specific, important features which are summarized below and which mark it out from other QTAIM software for the condensed phase (a list, not exhaustive, is given elsewhere [4, 5]). As an obvious prerequisite, application of QTAIM to the solid state implies a knowledge of at least the electron density and its derivatives in a representative portion of the system. If the system is periodic in nature this representative portion corresponds to the unit cell or, simply, to the unique part of it, the asymmetric unit. The usual practical implementations of QTAIM to solids usually work on a user’s defined volume, which includes the basin of the unique atoms within the system and the basins of their bonded atoms. Precise information about the periodic nature of the system is usually lost this way, and application of QTAIM to periodic systems is essentially brought back to a topological study of the electron density of a large cluster of atoms extracted from the crystal. The electron density is either calculated analytically or simply given on a grid [4], whereas the r derivatives, in particular those of order greater than two, are only estimated numerically by the large majority of software. (Topological analysis of rðrÞ and of ‘2rðrÞ requires rðrÞ derivatives up to the second and up to the fourth order, respectively.) A full implementation of QTAIM [6] also necessitates a knowledge of the one-electron density matrix (ODM) and of the main diagonal of the two-electron density matrix, the pair density; neither of these is usually available in QTAIM software for solid systems, however.
Most of the listed shortcomings are, on the contrary, simply absent from TOP- OND-98 [1, 7], because of its intimate interface with the libraries of, and output from, the CRYSTAL-98 package [8]. (TOPOND-98 is currently interfaced with CRYSTAL-98. Linking to CRYSTAL-2003, the most recent release of CRYSTAL, is currently in progress and planned to be complete by the end of 2006.) The CRYSTAL software performs ab-initio calculations of the ground-state energy, electronic wave function, and properties of periodic systems in 0 (molecules), 1 (polymers), 2 (slabs), and 3 dimensions (crystals). Systems with di erent periodicity are treated on an equal footing in CRYSTAL, with the single particle wave functions being expanded, for any periodicity, as a linear combination of Bloch functions, each of which is defined in terms of local atomic orbitals. Space symmetry is fully exploited, with 230 space groups, 80 layer groups, 99 rod groups, and 45 point groups available to the user. These unique features of CRYSTAL automatically make TOPOND a powerful tool for application of QTAIM to

7.2 QTAIM Applied to Solids – the TOPOND Package 167
molecules, polymers, surfaces, and crystals, using a single software product. TOPOND works on electron densities obtained with similar accuracy for di erent aggregations of matter, and topologically analyzes these densities with the same kinds of algorithm and precision. For example, one may very easily assess how the topological and atomic properties of a molecule are modified when it becomes surrounded by equivalent molecules in a given crystalline arrangement, or how the bonding patterns and atomic properties of a bulk atom in a solid change when this atom is placed on, and forms the surface of, a solid.
The intimate interface with CRYSTAL enables TOPOND to take advantage of the full periodic geometrical machinery of the former software. For example, this implies that when a critical point (CP) is located in a crystal, a full list of neighboring atoms, with their exact cell locations and with their coordinates given in fractional or Cartesian form, is immediately available. Or that all atoms of equivalent symmetry are easily recognized and unnecessary calculations are avoided as much as possible.
The electron density rðrÞ, [2, 9]:
XX
rðrÞ ¼ Pgm; nlwmgðrÞwlnðrÞ ð1Þ
g; l m; n
and, if needed, all its derivatives up to the fourth order are computed analytically by TOPOND. In Eq. (1) wm and wn are atomic orbitals, g and l are lattice vectors, Pg 1 is the ODM associated to orbitals located in crystal cells having relative position g l, and wmg is an atomic orbital located in cell g, but with same shape and the same fractional coordinates as the wm orbital in the reference zero cell (g ¼ 0). Evaluation of rðrÞ is not performed using a single threshold distance criterion from the point r to select whether a wmgðrÞ orbital contribution is to be included or not into the quadruple sum yielding rðrÞ. Instead, di erent distance thresholds are used for each orbital, based on the magnitude of their value at r [9]. This means that the ‘‘cluster of atoms’’ built around r for evaluating rðrÞ does not have a predetermined fixed size as in all other QTAIM implementations for solids, but a di erent size for each m, n orbital, reflecting its specific di useness. ODMs are available within TOPOND and quantities such as the kinetic energy densities K(r) or G(r), the virial density VðrÞ, or the electron localization function (ELF) [10] may be all evaluated exactly, without resorting to their approximate expressions [11] in terms of rðrÞ, ‘rðrÞ, and ‘2rðrÞ. Unfortunately, the Fermi hole and the ensuing localization/delocalization indices [12] are not yet computed by TOPOND, but their evaluation will, hopefully, be included in a future release of the software, at least for HF or Kohn–Sham type wavefunctions.
The TOPOND package presently comprises five sections [1, 3]. The first two implement topological analysis of rðrÞ and of ‘2rðrÞ, respectively. The fourth section of TOPOND deals with evaluation of atomic basin boundaries and associated atomic basin properties and the last section performs grid evaluation of several scalar fields, including the ELF, and traces out molecular/crystal graphs or, generically, ‘r trajectories in selected molecular/surface/crystal planes. The third sec-

168 7 Solid State Applications of QTAIM and the Source Function
tion, which is currently being implemented and not yet released is concerned with evaluation of IAS properties. These include, among others, the integrated surface charge, the net flux of the total electric field, yielding qðWÞ via the Gauss theorem [13], and the external source function contribution to the density within an atomic basin [14]. Two di erent CP search algorithms are available, the conventional Newton–Raphson (NR) technique and an eigenvector following (EF) method, proposed by Popelier [15]. The EF method can be seen as an NR method with a suitable and locally defined shift for the NR step. It is thus much less sensitive than the NR method to the choice of good starting search points. The EF method can seek CPs of a given kind, irrespective of the structure of the Hessian of the scalar field at the starting search point – a feature that makes it particularly helpful for topological study of ‘2r, because this function varies quite rapidly. Separate searches for the di erent kinds ð3; 3; 3; 1; 3; þ1; 3; þ3Þ of CPs are implemented in TOPOND for both the r and the ‘2r fields. A fully automatic search strategy, able to find sequentially all kinds of electron density CPs, is also available. This strategy makes use of the relevant EF step prescription for each kind of CP searched for in sequence. A CP search on a grid, defined in the asymmetric unit, may be also exploited when the Morse topological relationship [16], given by Eq. (2):
n b þ r c ¼ 0 |
ð2Þ |
(where n, b, r and c are the total number of nuclear, bond, ring and cage CPs) is not fulfilled by the set of CPs found using the fully automatic search. The CP search on a grid is usually found to be 2 to 3 orders of magnitude more demanding computationally than the automatic search [3]. If one adopts suitable grid sizes, however, it seldom misses CPs, even when very flat density distributions occur, as in the metals. Atomic interaction lines (r field) and atomic graphs (‘2r field) are determined by TOPOND by tracing the associated steepest ascent/ descent ‘r or ‘ð‘2rÞ paths using a fifth-order Runge–Kutta method with monitoring of local truncation error and adaptive step-size control. Correct parameterization of the algorithm (desired accuracy at each step, initial step size, oscillation control close to the 3, 3 attractors) enables tracing of the correct atomic interaction lines for metals also; for these the occurrence of nonnuclear attractors (NNAs) [17] is more the rule than the exception and the network of interaction lines is, consequently, rather complicated and largely unstable with regard to changes in the computational model or in the cell parameters [1, 3, 18, 19].
7.2.1
QTAIM Applied to Experimental Densities: TOPXD and XD Packages
During the last decade, QTAIM has increasingly been applied to crystalline systems [20, 21]. This is, on the one hand, because of the technical developments that have made X-ray di raction a unique tool for mapping the charge density in

7.2 QTAIM Applied to Solids – the TOPOND Package 169
crystals and, on the other hand, because of noticeable improvements in ab-initio periodic approaches [22, 23], which have enabled calculation of reliable electron densities even for crystals having a large number of atoms (>50–100) in the unit cell. One may safely say that QTAIM is the primary standard theory used by the X-ray density community to discuss bonding in crystals [21]. This would not have been possible if software packages implementing the QTAIM for experimental densities had not been developed, documented, and made generally available in the past decade. One such software package is the TOPXD program [24, 25], which enables complete topological analysis of experimental charge densities on the basis of the Hansen–Coppens multipole formalism [26]. It is based on TOPOND-98, but with subroutines for geometrical calculation and density evaluations rewritten in the XD package convention [27, 28]. XD is the most widely distributed package for experimental charge density multipole refinement and TOPXD is fully integrated in the most recent version, XD 5.01, [27, 28]. The main features of TOPXD are those of TOPOND, with more extended documentation, friendly input style, increased speed in evaluating the IAS, and added facilities for their 3D visualization. The experimental electron density and its derivatives up to order 2 are calculated analytically, whereas derivatives of third and fourth order are obtained with great precision as a numerical finite-di erence approximation of the first and second-order analytical derivatives [24]. In contrast with TOPOND, properties at a given point r are calculated by including density contributions from ‘‘pseudoatoms’’ which lie within a given distance threshold of r. This may limit the accuracy of the calculated properties and requires a check that the properties being computed converge relative to the distance threshold [27]. The ODM is not available within TOPXD and XD and all properties depending on this matrix (Section 7.2) can clearly not be computed.
One of the most important reasons for the popularity of QTAIM is that a large part of this theory uses, operationally, only information contained in the electron density r(r), which enables unbiased comparison of theoretical and static experimental densities, irrespective of their diverse origin and of the di erent approximations one makes to derive them [20, 21, 29]. Such a comparison may provide information about the quality of experimental data and the suitability of the multipolar model used to project the reciprocal space representation of r to its real-space counterpart [30, 31]. Conversely, it may reveal deficiencies of the theoretical approach [30–32], for example poor treatment of the electron correlation, the use of an insu ciently flexible basis set, or the adoption of a pseudopotential with core–valence separation which is too crude.
The performance of the adopted multipolar model may be tested as follows [24, 31, 33]. First, a multipole refinement, keeping the positional parameters fixed and thermal parameters set to 1, is performed using structure factors F generated with an ab-initio periodic calculation to simulate the X-ray di raction data set. Topological analysis is then performed on the density from the multipole model and the results compared with those obtained using the primary theoretical density. It has been shown that the multipole model may significantly bias the topolog-

170 7 Solid State Applications of QTAIM and the Source Function
ical results, because of the limited flexibility of the radial functions used in the multipolar analysis. Indeed, the observed discrepancies between theoretical and experimental, X-ray-derived topological properties are usually found to decrease substantially when the theoretical densities are projected into the multipole density functions by refinement of the theoretical F. All of this analysis may be performed easily by using CRYSTAL, TOPOND, and TOPXD/XD software in combination.
7.3
QTAIM Applied to Molecular Crystals
Electron distributions of crystals are an amazing source of information about the weaker and the less conventional atomic interactions [21], besides that provided about standard chemical bonds. Typical of molecular crystals is the simultaneous occurrence of normally strong intramolecular bonds, and of generally weak intermolecular contacts, with properties of both kinds of interaction being mutually a ected, in contrast with the situation for the isolated molecule or the simple case of gas-phase molecular aggregates (dimers, trimers, . . .). QTAIM has proved to be a very powerful tool for isolating, detailing the weak intermolecular interactions responsible of molecular crystal formation and for quantitative characterization of the e ect of these interactions on intramolecular bonding.
Using the urea crystal as an example, answers will be provided to several simple questions:
1.How important are packing e ects on intramolecular bonds?
2.Does the packing have di erent impact on the di erent atoms/chemical groups present in the molecule?
3.How large is the enhancement of the molecular dipole on crystallization?
4.How can each oxygen atom in the urea crystal be involved in four NaH O hydrogen bonds (HBs)?
5.How does the global molecular volume contraction observed in the solid result from the individual atomic volume change on crystallization?
Before illustrating the situation for urea, it is worth mentioning that much more complex QTAIM applications to molecular crystals have appeared. To save space, however, these studies cannot be discussed here. I merely quote Refs [34] and [35] that address the interesting problem of the nature and function of the weak CH O intermolecular interactions in crystals by analyzing the experimental and theoretical densities of the 3,4-bis(dimethylamino)-3-cyclobutene-1,2-dione (DMACB) crystal. This system is characterized by 23 unique intermolecular and intramolecular CH O interactions and by no other type of stronger, and thus successfully competing, HB. References [34] and [35] discuss basic questions such as:
7.3 QTAIM Applied to Molecular Crystals 171
1.Does the existence or the absence of an CH O CP reflect specific geometrical features of a CH O contact in the DMACB crystal?
2.Can the bonded CH O contacts in this crystal be classified as true HB?
3.Do the weak intermolecular HBs induce a large molecular dipole moment enhancement on crystallization, as typically found in molecular crystals tied by the much stronger NH O and OH O bonds?
4.Do the crystal and procrystal densities di er in the topological features of their CH O contacts and can the CH O bond energies thus be reliably retrieved from the BCP properties alone?
The reader is referred to the original papers to discover why three ‘‘YES’’ and one ‘‘NO’’ were the answers to these questions.
7.3.1
Urea
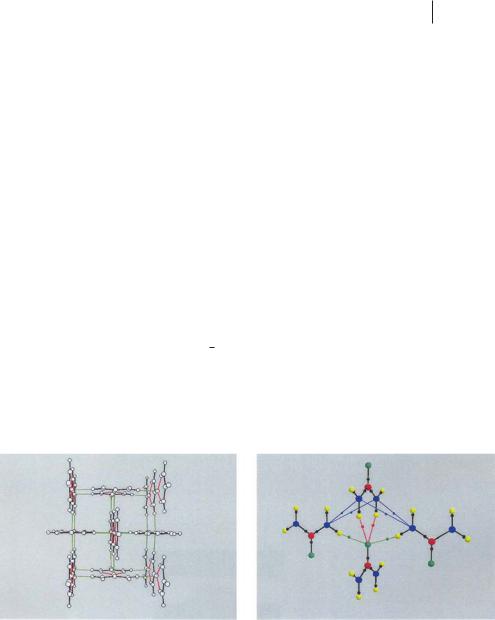
In the crystal structure (space group P421m), the urea molecules are linked to each other through HBs to form infinite planar tapes (OaH00 length 2.06 A˚ , HBs shown in red in Fig. 7.1) [36]. Adjacent tapes are mutually orthogonal and oriented in opposite directions, their cohesion being provided by another set of HBs (OaH0 length 1.99 A˚ , shown in green in Fig. 7.1). Each oxygen atom is in-
Fig. 7.1 The urea crystal. Left: The view is approximately along the c axis. Molecules are linked to each other through HBs (red lines) to form infinite planar tapes. Adjacent tapes are mutually orthogonal, oriented in opposite directions and tied together by another set of HBs (green lines). Right: Intramolecular and intermolecular bond paths (BCPs: small dots)
for an urea molecule linked through HBs with another molecule along a tape and with two molecules of two neighboring orthogonal tapes. Bond paths in blue are associated with the shorter NaN contacts (see text). Each oxygen atom is involved in four HBs, two within the planar tape and two with neighboring orthogonal tapes.