

1. Ключевые открытия, сделанные в результате анализа генома человека
Размер гаплоидного генома человека составляет 2858 млн нуклеотидных пар. Он содержит примерно 22 585 генов, кодирующих разнообразные белки. Эта цифра несколько завышена, так как среди открытых рамок считывания, предсказанных компьютерными программами, могут встречаться и псевдогены. Если мы удалим из этого множества генов все транскрипты псевдогенов, в которых ученые уверены не полностью, останется 18 349 кодирующих участков. Такая оценка несколько занижена, поскольку некоторые предсказанные транскрипты могут оказаться еще не описанными генами, которые встречаются только у человека. Таким образом, мы пришли к выводу, что у человека настоящих генов от 18 000 до 22 000. Интересно, что это число оказалось намного меньше, чем считалось в догеномные времена.
По первоначальным оценкам, число генов у человека было в пределах от 50 тыс. до 150 тыс. (рис. 1).Точная оценка числа генов у человека пока невозможна по многим причинам. Гены, расположенные в некоторых хромосомных участках, трудно поддаются учету. К числу таких участков относятся псевдоаутосомные районы — гомологичные локусы, расположенные на концах X- и Y-хромосом и способные к конъюгации и кроссинговеру. По одним методам подсчета, гены, находящиеся в этих районах, учитываются один раз, а по другим — два. Подобные трудности представляют собой дуплицированные гены и тандемные генные кластеры. Длина таких кластеров может различаться у разных людей, и рекомбинация между ними приводит к появлению кластеров с измененным числом тандемных генных повторов, что вызывает появление дополнительных копий того или иного гена и вносит дополнительные неясности в определение числа генов у человека.
Было выявлено, что некоторые довольно протяженные участки генома вообще отсутствуют у целого ряда вполне здоровых людей. Длина таких «необязательных» участков может составлять от нескольких тысяч до нескольких миллионов нуклеотидных пар. В настоящее время описано около 2000 таких участков, получивших название «вариации числа копий» (сору number variation, или CNVs). При этом на каждый индивидуальный геном приходится более 100 таких вариаций со средним размером 250 тыс. пар нуклеотидов каждая. Для сравнения: ген человека в среднем занимает около 60 тыс. пар нуклеотидов. Нередко внутрь такого вариантного участка попадают цельные гены, это означает, что у отдельно взятого человека какой-либо ген может быть представлен не двумя, а одной или, наоборот, тремя копиями, что приводит к изменению количества данного гена.
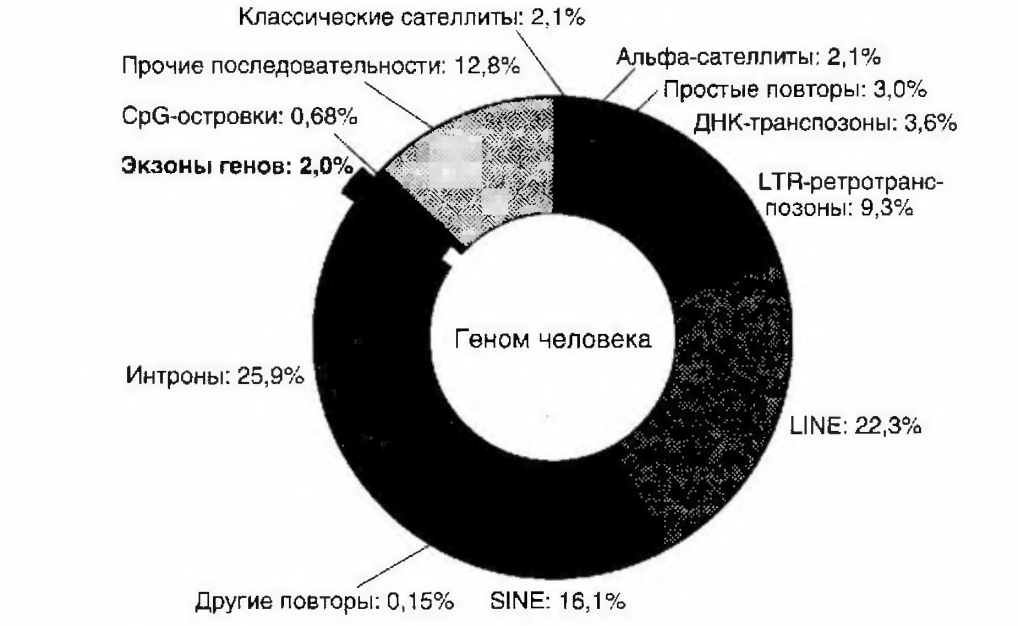
Рисунок 3- Содержание нуклеотидных последовательностей (НП) в геноме человека. НП, кодирующие белки (примерно 20 тыс. генов), занимают менее 1,5% генома [1]
В некоторых случаях ген вообще может быть полностью утрачен без существенного изменения фенотипа. Как вариации числа копий, так и другие непатологические геномные особенности вносят свой вклад в уникальность и каждого генома, и каждого человека. Но не все вариации числа копий абсолютно безвредны. Только для заболеваний нервной системы в настоящее время описано 17 CNV, приводящих к возникновению различной патологии, в том числе к болезни Альцгеймера или Паркинсона, аутизму, шизофрении. Для учета новой информации, касающейся вариаций числа копий в норме и в патологии, создана специальная база данных DECIPHER.
Все кодирующие участки ДНК составляют не более 1,5% длины всего генома. Остальное пространство заполнено некодирующими РНК, роль которых до сих пор не ясна, регуляторными последовательностями, а также повторяющимися участками ДНК, многие из которых являются производными различных вирусов и эндогенных самореплицирующихся молекул. Стоит отметить, что, несмотря на малую долю, занимаемую кодирующими участками, примерно 80% всего генома с той или иной скоростью транскрибируется, просто многие транскрипты не транслируются в белки. В первую очередь, это — последовательности интронов, вырезаемых при сплайсинге, множественные рибосомные РНК, транспортные РНК, а также многочисленные малые функциональные РНК, такие как микроРНК. Некодирующие РНК явно потеснили белки на их пьедестале главных молекул, обеспечивающих жизнедеятельность клеток. Скорее всего, мир некодирующих РНК только начал открываться исследователям, и в полной мере значение таких РНК можно будет оценить лишь в будущем.
Большую часть некодирующих РНК человека составляют РНК «домашнего хозяйства» клетки, непосредственно не участвующие в синтезе белка. К таким РНК относят структурные рибосомные РНК (рРНК), транспортные РНК (тРНК), 7SК РНК, РНК, входящие в состав сплайсосом, и другие. В геноме человека гены, кодирующие рРНК и тРНК, представлены многочисленными копиями — около 500 генов, кодирующих тРНК, и около 200 копий для рРНК. Гены, кодирующие рРНК, расположены на коротких плечах 5 акроцентрических хромосом (13, 14, 15, 21, 22).
Важным открытием последнего времени стало обнаружение в геноме человека «нетрадиционных» РНК-транскриптов, не способных кодировать белок, но способных модулировать функции белковых продуктов, считываемых с других генов, а также их пространственное распределение в клетке. Такие РНК называют риборегуляторами. Например, некодирующая РНК Н19 имеет отношение к целому ряду процессов, протекающих в клетках, в том числе к запуску клеточного цикла, а также играет важную роль при злокачественном перерождении клеток.
Локус HFE, участвующий в метаболизме железа и отвечающий за наследственное заболевание гемохроматоз, кодирует не только мРНК для белка HFE, но и антисмысловую РНК, образующуюся на другой нити ДНК и регулирующую мРНК. Этот антисмысловой (комплементарный) РНК-продукт способен взаимодействовать с основной мРНК HFE, образуя РНК-РНК гибриды, неспособные транслироваться в рибосомах с образованием белка. Еще один интересный ген — SRA, кодирующий РНК-активатор стероидного рецептора. Он обеспечивает активность стероидных рецепторов за счет образования комплекса с этим белком.
Важность клеточных функций некодирующих РНК подтверждается наблюдением, что их промоторы более консервативны, чем промоторы генов, кодирующих белки. В клетках человека также выявлены короткие двунитевые РНК (микроРНК) размером 21-32 нуклеотидные пары, участвующие в процессе регуляции и экспрессии генов путем РНК-интерференции. Этот механизм впервые был обнаружен в 1998 г. у низших животных, в том числе нематоды С. elegans. Оказалось, что микроРНК человека также способны уменьшать работу строго определенных генов путем воздействия на процесс синтеза кодируемых ими белков. Подавление экспрессии может происходить как на уровне транскрипции, так и посттранскрипционно. У человека число экспериментально подтвержденных микроРНК превышает 700. По разным оценкам, от 10 до 30% всех генов белков регулируется микроРНК.
Около 45% генома человека составляют мобильные генетические элементы, большая часть которых является ретротранспозонами, использующими РНК в жизненном цикле, и только меньшая часть (3%) представлена ДНК- транспозонами. Ретротранспозоны делятся на короткие (SINE), длинные (LINE) и длинные концевые повторы (LTR). Элементы SINE занимают 13% длины генома, основным типом этих элементов являются специфические для человека ALU-повторы, они составляют 10% общей длины человеческого генома. Следующий класс транспозонов — LINE-повторы, составляет 21% длины генома, в этом классе также выделяется преобладающий элемент — L1, он занимает 17% длины генома. LTR-транспозоны — самые малочисленные, они занимают только 8% общей длины генома.
Известно, что многие ретротранспозоны способны транскрибироваться, в том числе в различные малые РНК. При стрессовых воздействиях (тепловой шок, вирусная инфекция и др.) уровень малых РНК, транскрибирующихся с SINE, разбросанных по геному, увеличивается многократно. Эти малые РНК важны для выживания клеток в неблагоприятных условиях. Кроме ретротранспозонов и ДНК-транспозонов, в геноме представлены и другие типы повторяющихся последовательностей. Примерно 0,1% общей длины генома занимают простые повторы, например ЦАЦАЦАЦАЦАЦАЦА.
Псевдогены также считаются повторами. Псевдогенами называют последовательности, родственные генам, но лишенные способности кодировать белки. Как правило, псевдогены не имеют промотора и интронов. Псевдогены, в которых отсутствуют интроны гена-предшественника, называют процессированными псевдогенами. Предполагается, что процессированные псевдогены возникают путем интеграции в геном комплементарной ДНК (кДНК), образовавшейся в результате обратной транскрипции соответствующей мРНК, перед этим претерпевшей полный сплайсинг. Встраивание такой кДНК может быть обеспечено с помощью белковых продуктов, кодируемых автономными ретротранспозонами.
Многие псевдогены человека сохраняют очень большое сходство с функциональными родственниками. Сходство между псевдогенами и родственными им генами может достигать от 40 до 100%. Некоторые псевдогены весьма похожи на гены: они имеют промотор, ГЦ-островки, интроны и сайты сплайсинга, однако функционировать не могут. Нарушение функции такого псевдогена, как правило, происходит в результате мутации, в частности вводящего стоп-кодон в середину открытой рамки считывания или сдвиг рамки. Иногда эти псевдогены утрачивают и способность к транскрипции, но чаще их мРНК считывается, но не транслируется.
Разные хромосомы человека содержат различное количество генов. Эта величина зависит не только от размера хромосомы, но и от плотности генов на ней. Самое большое число генов находится на первой хромосоме (2316 генов), самое маленькое — на Y-хромосоме (94 гена). Первая хромосома — самая длинная, но плотность генов самая большая не на ней (10,29), а на 19-й хромосоме — в среднем 26 генов на каждый миллион пар оснований длины. На втором месте 17 хромосома с плотностью генов 16,2. Плотность генов в каждом конкретном участке ДНК зависит от содержания гуанина и цитозина: чем больше на участке ГЦ-пар, тем больше на нем генов.
Как и у всех эукариот, гены человека состоят из экзонов и интронов. Экзонами называют участки ДНК, остающиеся в составе мРНК после завершения сплайсинга, а интронами — участки ДНК, транскрибируемые в РНК, но вырезаемые при сплайсинге, а значит, отсутствующие в составе зрелой информационной РНК, служащей матрицей для биосинтеза белка. Интроны, как правило, начинаются с последовательности ГУ и заканчиваются на АГ. Один и тот же первичный РНК-транскрипт может быть сплайсирован в целый ряд зрелых мРНК, различающихся по своему экзонному составу. Таким образом, один и тот же участок ДНК может служить экзоном или входить в состав интрона, в зависимости от того, какой именно сценарий сплайсинга реализуется в данной клетке.
Процесс реализации различных тканеспецифичных сценариев сплайсинга называют альтернативным сплайсингом (рис. 2). Доказано, что у человека 92-94% генов подвержено альтернативному сплайсингу; при этом у 86% генов дополнительные изоформы составляют не менее 15% общего количества мРНК, синтезируемой с данного гена. Интересно, что геном круглого червя С. elegans по количеству генов практически не отличается от генома человека, однако альтернативному сплайсингу подвергаются пре-мРНК только 15% генов этой нематоды. Видимо, основное назначение альтернативного сплайсинга — увеличение разнообразия белковых продуктов, считываемых с генов, без пропорционального увеличения числа самих генов и размера генома. Альтернативный сплайсинг также позволяет осуществлять сложную тканеспецифичную систему регуляции экспрессии генов человека.
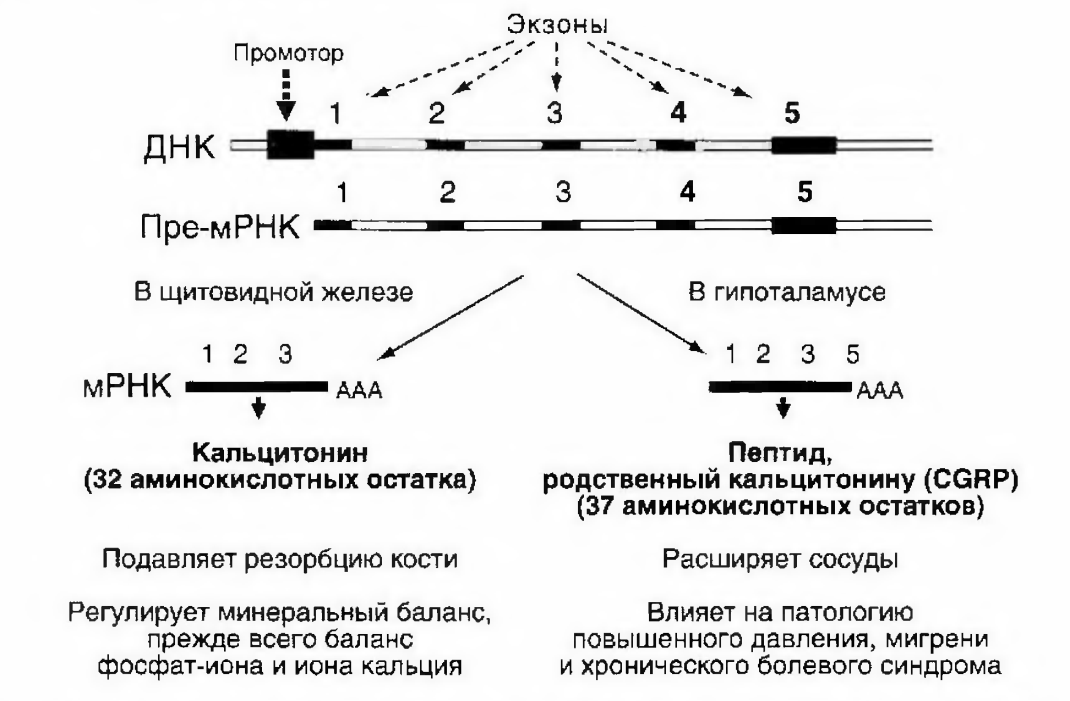
Рисунок 4 – Пример альтернативного сплайсинга гена CALC1, кодирующего 2 разных гормона – кальцитонин и родственный ему пептид. Кодирование достигается путем использования двух альтернативных экзонов 4 и 5 [1].
У большинства генов, не подверженных альтернативному сплайсингу, интронов вообще нет — они представляют собой единый цельнотранскрибируемый экзон. Таких генов у человека 751, при этом длина этого единственного экзона значительно выше средней длины экзона для многоэкзонных генов, в среднем на один ген человека приходится по 9 экзонов и 8 интронов. Более 80% экзонов на каждой хромосоме не превышают в длину 200 пар азотистых оснований, средняя длина экзона — 170 пар. Интроны также редко бывают менее 20 пар оснований, таковых меньше 0,01% всех интронов. Менее 10% интронов превышают в длину 11 тыс. пар. Возможно, это связано с трудностями сплайсинга слишком коротких и слишком длинных фрагментов. Интересно, что чем длиннее хромосома, тем длиннее на ней интроны и межгенные участки ДНК, коэффициент корреляции составляет 0,95 для интронов и 0,97 для межгенных участков.
Обнаружены отдельные локусы, где внутри одного гена целиком содержится другой, «вложенный» ген. Как правило, при этом типе организации один белок-кодирующий ген располагается в интроне другого белок-кодирующего гена. Но встречаются и другие варианты. В качестве примера можно привести митохондриальный ген одной из рибосомных РНК. Ген, кодирующий данную рРНК, обеспечивает ею рибосомы митохондрий в качестве структурного компонента (т.е. не кодирует белок). Но вместе с тем небольшой участок, расположенный внутри этого гена, кодирует короткий белок, получивший название «гуманин» (от англ. human — человек), который принимает участие в процессе программированной клеточной гибели. В этом случае белок-кодирующий ген заключен внутри РНК-кодирующего гена. Другой вариант — уже упоминавшийся выше ген Н19. Здесь, наоборот, ген IGF2, кодирующий белок, содержит внутри своей кодирующей части другой более короткий ген, кодирующий только РНК, которая принимает участие в регуляции работы этого гена.
Средняя длина типичного белок-кодирующего гена составляет 16995 нуклеотидов, если же не учитывать неизученные пока транскрипты, предсказанные компьютером, длина среднего белок-кодирующего гена составит 21 461 нуклеотид. Очень длинные гены, длиннее 250 тыс. пар оснований, составляют всего 4% набора генов человека. Самый длинный из генов человека – ген CNTNAP2 (2,3 Мб) – это ген белка Caspr 2. На втором месте ген DMD миодистрофина – его длина 2,22 Мб.
Многие гены человека объединены в генные семейства. Гены объединяют в семейства в случае, если их экзоны родственны между собой, т.е. похожи по нуклеотидной последовательности; в геноме человека присутствует около 1500 таких семейств генов. Причем только около сотни из них специфичны для человека и других позвоночных животных, тогда как основная масса генных семейств намного более консервативны — они имеются и у человека, и у других многоклеточных.
Гены, принадлежащие к одному семейству и встречающиеся у одного организма, называют паралогичными; одинаковые гены, присутствующие у разных видов, — ортологичными. Как правило, паралогичные гены эволюционировали путем дупликации и последующего расхождения (дивергенции) копий одного гена-предщественника. Одна из дуплицированных копий гена может приобретать новые функции, в то время как другая обеспечивает поддержание исходных. Такие дупликации в процессе эволюции происходили неоднократно. Подсчитано, что в геноме человека в сумме дуплицировано около 3,6% нуклеотидных последовательностей размером в 1000 пар нуклеотидов и более. Разные копии одного семейства генов могут располагаться в геноме рядом и следовать друг за другом (это называют тандемной дупликацией) или находиться в разных хромосомах.
Функции ортологичных генов консервативны. Например, ген Рах6 имеет одинаковую функцию у человека, дрозофилы, головоногих моллюсков и нематоды. Этот транскрипционный фактор необходим для развития органов зрения у всех билатерально-симметричных животных, включая человека и других млекопитающих. Если ген Рах6 человека эктопически экспрессировать в организме дрозофилы, это приведет к появлению у нее дополнительных глаз, что указывает на эволюционную консервативность всей нижележащей генной сети. Сходны и фенотипические эффекты мутаций Рах6 у различных организмов, они всегда проявляются повреждением органов зрения. У человека гетерозиготы по мутации в Рах6 страдают аниридией. У мыши этот же фенотип был описан как «small eyes» — уменьшенные и упрощенные глаза. Гомозиготы по мутации Рах6 не доживают до рождения ни у мыши, ни у человека, такие эмбрионы страдают серьезным дефектом развития головного мозга, носа и ушей.
Как правило, ортологичные транскрипционные факторы более сходны между собой, чем структурные белки. Этот феномен и называют эволюционной консервативностью. Однако консервативность не мешает транскрипционным факторам образовывать крупные генные семейства, например семейство генов Нох.
Функции дивергировавших генов одного семейства (паралогичных) чаще всего остаются сходными, но экспрессия членов такого семейства, как правило, осуществляется на разных стадиях развития организма человека или в разных типах клеток. Так, в геноме человека обнаружено 22 гена, кодирующих родственные факторы роста фибробластов, — все они структурно сходны друг с другом и подразделяются на 6 подсемейств. При этом у низших организмов число таких генов существенно меньше, например, у дрозофилы их всего два. Другой пример — гены, кодирующие кератины — белки наружного слоя кожи и ее производных (волосы, ногти). Их у человека 54. Правда, большинство из этих генов сконцентрировано в составе генных кластеров на хромосомах 12 и 17.
Интересно, что некоторые генные семейства у человека представлены меньшим числом генов, чем у низших животных. Например, геном нематоды С. elegans содержит 25 генов, кодирующих белки-иннексины, образующие щелевые каналы, а геном человека — только три родственных иннексинам гена паннексинов. Подобным примером могут послужить обонятельные рецепторы, представленные в геноме человека более чем 1000 копий. Но примерно 60% из них являются псевдогенами, что может объяснять относительно плохо развитое чувство обоняния у человека, у мыши примерно из такого же числа генов, кодирующих рецепторы обоняния, работает не менее 80%.
Считается, что массовая потеря функциональности генов обонятельных рецепторов у вида Homo sapiens произошла за последние 6 млн лет, прошедших с момента обособления семейства гоминид от их ближайших родственников — человекообразных обезьян. У человекообразных обезьян доля функциональных генов в семействе обонятельных рецепторов выше, тем не менее и у них около 30% данных генов представлены псевдогенами. Такую потерю функциональности генов обонятельных рецепторов связывают со снижением роли обоняния в жизни человека и человекообразных обезьян по сравнению с другими млекопитающими и переходом роли ведущего анализатора к зрительному, после появления у приматов трехмерного цветного зрения.
Завершение работы над последовательностью генома человека прояснило многие вопросы эволюции. Например, оказалось, что геномы человека и шимпанзе различаются примерно на 1,23%. Около 0,25% пар нуклеотидов, различающих геномы человека и шимпанзе, приходятся на полиморфные состояния ДНК, часто встречающиеся и в популяциях человека. Значит, на долю истинных различий между обезьяной и человеком остается менее 1%. Но на проблему сходства и различия человека и шимпанзе можно посмотреть и по-другому — учитывая не общую длину последовательности генома и процент от нее, а отдельные гены.
Такое сравнение показало, что примерно 6% всех функциональных генов уникальны либо для человека, либо для шимпанзе. Другими словами, наиболее значимая разница между этими видами заключается не в точечных мутациях ДНК, а в геномных перестройках, приведших к изменению числа генов в составе генных семейств, а также изменению функции и спектра экспрессии некоторых генов. В среднем ген человека отличается от ортологичного гена шимпанзе на две аминокислотных замены. Примерно треть генов человека и шимпанзе на уровне белка вообще неотличимы. Самое главное различие между двумя родственными геномами — это человеческая хромосома 2, образовавшаяся в результате слияния 12-й и 13-й хромосом шимпанзе, впоследствии переименованных в хромосомы 2А и 2В.
Большое значение расшифровка генома человека имела и для понимания эволюции человека как вида. Международная программа НарМар, в которой принимают участие исследовательские группы и фонды из США, Великобритании, Канады, Японии, Китая и Нигерии, создала базу данных по гаплотипам, встречающимся в популяциях европейского, азиатского и африканского происхождения. Гаплотип — совокупность генетических маркеров, расположенных рядом и наследующихся вместе на одной хромосоме. В качестве таких маркеров, благодаря возросшей эффективности секвенирования, стало возможным использовать SNP (Single Nucleotide Polymorphisms) — однонуклеотидные полиморфизмы. Большинство однонуклеотидных полиморфизмов нейтральны, поэтому их распространение в популяциях не подвержено влиянию естественного отбора и позволяет проследить, в частности, миграции своих носителей — людей, составляющих популяции. Благодаря успехам программы НарМар была подтверждена гипотеза общей африканской прародины человечества, причем Африка оказалась более разнообразной по гаплогруппам Y-хромосомы, чем весь остальной мир. Подобные результаты дали и работы по митохондриальной ДНК — разнообразие митотипов в Африке оказалось существенно больше, чем во всем остальном мире. Таким образом, гипотеза замещения евразийских популяций архаичного неандертальца популяциями современного человека, пришедшими из Африки, получила мощную поддержку.
По мере расселения человека за пределы Африки частоты древних гаплогрупп изменялись благодаря генному дрейфу, эффекту основателя, естественному отбору и другим причинам, в результате чего в разных популяциях современного человека частоты гаплогрупп также различаются.
Постепенно в результате мутаций в некоторых областях появлялись и новые гаплогруппы. В этих областях такие «молодые» гаплогруппы часто ассоциированы с различными генетическими заболеваниями, что позволяет проводить их относительно быструю диагностику, т.к. для поиска мутации не требуется типирование всего генома пациента. Для каждой гаплогруппы, статистически ассоциированной с тем или иным распространенным заболеванием, достаточно определение одного маркера. Такой подход, предполагающий наличие часто встречающегося предрасполагающего генетического варианта, получил название CDCV (common decease-common variant). Именно он послужил основой проекта НарМар.
Стоит заметить, что на настоящий момент гипотеза CDCV пока не смогла окончательно победить конкурирующую концепцию CDRV (common decease -rare variant), которая предполагает, что распространенные заболевания, такие как ожирение, сердечно-сосудистые заболевания или сахарный диабет, возникают благодаря множеству редких аллелей, каждая из которых увеличивает шанс возникновения той или иной болезни. В связи с этим можно предположить, что различия между CDCV и CDRV не так велики. Возможно существование как нескольких распространенных гаплотипов, вносящих небольшой вклад в развитие диабета, так и большого количества редких аллелей, по отдельности вносящих гораздо больший вклад.
Клиническое значение данных, полученных в проекте НарМар, пока меньше теоретических, однако разработанные в ходе этого проекта новые подходы, такие как полногеномный анализ ассоциаций, внушают сдержанный оптимизм.