152 Chapter 5 ● Proteins: Their Biological Functions and Primary Structure
among other products, two dipeptides, each of which contained C-terminal isoleucine. One of these dipeptides migrated as an anionic species upon electrophoresis at neutral pH.
d. The tryptic tripeptide was degraded in an Edman sequenator, yielding first A, then B:
|
|
|
O |
H |
H |
|
|
|
C |
A |
A |
|
|
|
COCOCH3 |
|
|
|
|
|
A. |
|
N |
|
A |
|
|
|
|
|
|
C |
N |
CH3 |
|
|
|
i |
|
|
|
S |
|
H |
|
|
|
|
|
|
|
|
O |
H |
H |
|
|
|
|
|
|
|
C |
A |
A |
|
|
|
COCOCH2O CH3 |
|
B. |
N |
|
|
A |
|
|
|
C |
N |
CH3 |
|
|
|
i |
|
What is an amino acid sequence of the octapeptide? Four sequences are possible, but only one suits the authors. Why?
8. An octapeptide consisting of 2 Gly, 1 Lys, 1 Met, 1 Pro, 1 Arg, 1 Trp, and 1 Tyr was subjected to sequence studies. The following was found:
a. Edman degradation yielded
O H
C A
COH
N
N
C i
H
S
b.Upon treatment with carboxypeptidases A, B, and C, only carboxypeptidase C had any effect.
c.Trypsin treatment gave two tripeptides and a dipeptide.
d.Chymotrypsin treatment gave two tripeptides and a dipeptide. Acid hydrolysis of the dipeptide yielded only Gly.
e.Cyanogen bromide treatment yielded two tetrapeptides.
f.Clostripain treatment gave a pentapeptide and a tripeptide.
What is the amino acid sequence of this octapeptide?
9. Amino acid analysis of an oligopeptide containing nine residues revealed the presence of the following amino acids:
Arg Cys Gly Leu Met Pro Tyr Val
The following was found:
a.Carboxypeptidase A treatment yielded no free amino acid.
b.Edman analysis of the intact oligopeptide released
|
O |
H |
H |
|
C |
A |
A |
|
COCH2OCOCH3 |
|
N |
|
|
A |
|
C |
N |
CH3 |
|
i |
|
c.Neither trypsin nor chymotrypsin treatment of the nonapeptide released smaller fragments. However, combined trypsin and chymotrypsin treatment liberated free Arg.
d.CNBr treatment of the eight-residue fragment left after combined trypsin and chymotrypsin action yielded a six-residue fragment containing Cys, Gly, Pro, Tyr, and Val; and a dipeptide.
e.Treatment of the six-residue fragment with -mercap- toethanol yielded two tripeptides. Brief Edman analysis of the tripeptide mixture yielded only PTH-Cys. (The sequence of each tripeptide, as read from the N-terminal end, is alphabetical if the one-letter designation for amino acids is used.)
What is the amino acid sequence of this nonapeptide?
10. Describe the synthesis of the dipeptide Lys-Ala by Merrifield’s solid phase chemical method of peptide synthesis. What pitfalls might be encountered if you attempted to add a leucine residue to Lys-Ala to make a tripeptide?
Creighton, T. E., 1983. Proteins: Structure and Molecular Properties. San Francisco: W.H. Freeman and Co., 515 pp.
Creighton, T. E., ed., 1997. Protein Function—A Practical Approach, 2nd ed. Oxford: IRL Press at Oxford University Press.
Dayhoff, M. O., 1972–1978. The Atlas of Protein Sequence and Structure, Vols. 1–5. Washington, DC: National Medical Research Foundation.
Deutscher, M. P., ed., 1990. Guide to Protein Purification. Vol. 182, Methods in Enzymology. San Diego: Academic Press, 894 pp.
Goodsell, D. S., and Olson, A. J., 1993. Soluble proteins: Size, shape and function. Trends in Biochemical Sciences 18:65–68.
Heijne, G. von, 1987. Sequence Analysis in Molecular Biology: Treasure Trove or Trivial Pursuit? San Diego: Academic Press.
Hill, R. L., 1965. Hydrolysis of proteins. Advances in Protein Chemistry 20:37– 107.
Hirs, C. H. W., ed., 1967. Enzyme Structure. Vol. XI, Methods in Enzymology.
New York: Academic Press, 987 pp.
Hirs, C. H. W., and Timasheff, S. E., eds., 1977–1986. Enzyme Structure, Parts E–L. New York: Academic Press.
Hsieh, Y. L., et al., 1996. Automated analytical system for the examination of protein primary structure. Analytical Chemistry 68:455–462. An analyti-
cal system is described in which a protein is purified by affinity chromatography, digested with trypsin, and its peptides separated by HPLC and analyzed by tandem MS in order to determine its amino acid sequence.
Hunt, D. F., et al., 1987. Tandem quadrupole Fourier transform mass spectrometry of oligopeptides and small proteins. Proceedings of the National Academy of Sciences, U.S.A. 84:620–623.
Johnstone, R. A. W., and Rose, M. E., 1996. Mass Spectrometry for Chemists and Biochemists, 2nd ed. Cambridge, England: Cambridge University Press.
Karger, B. L., and Hancock, W. S., eds. 1996. Methods in Enzymology 271,
Section III: Protein Structure Analysis by Mass Spectrometry, J. R. Yates; Peptide Characterization by Mass Spectrometry, B. L. Gillece-Castro and J. T. Stults. New York: Academic Press.
Mann, M., and Wilm, M., 1995. Electrospray mass spectrometry for protein characterization. Trends in Biochemical Sciences 20:219–224. A review of the basic application of mass spectrometric methods to the analysis of protein sequence and structure.
Merrifield, B., 1986. Solid phase synthesis. Science 232:341–347.
Quadroni, M., et al., 1996. Analysis of global responses by protein and peptide fingerprinting of proteins isolated by two-dimensional electrophoresis. Application to sulfate-starvation response of Escherichia coli. European Journal of Biochemistry 239:773–781. This paper describes the use of tandem MS in the analysis of proteins in cell extracts.
154 Chapter 5 ● Proteins: Their Biological Functions and Primary Structure
Semipermeable bag containing protein solution
 Dialysate
Dialysate
Stir bar
Magnetic stirrer for mixing
FIGURE 5A.2 ● A dialysis experiment. The solution of macromolecules to be dialyzed is placed in a semipermeable membrane bag, and the bag is immersed in a bathing solution. A magnetic stirrer gently mixes the solution to facilitate equilibrium of diffusible solutes between the dialysate and the solution contained in the bag.
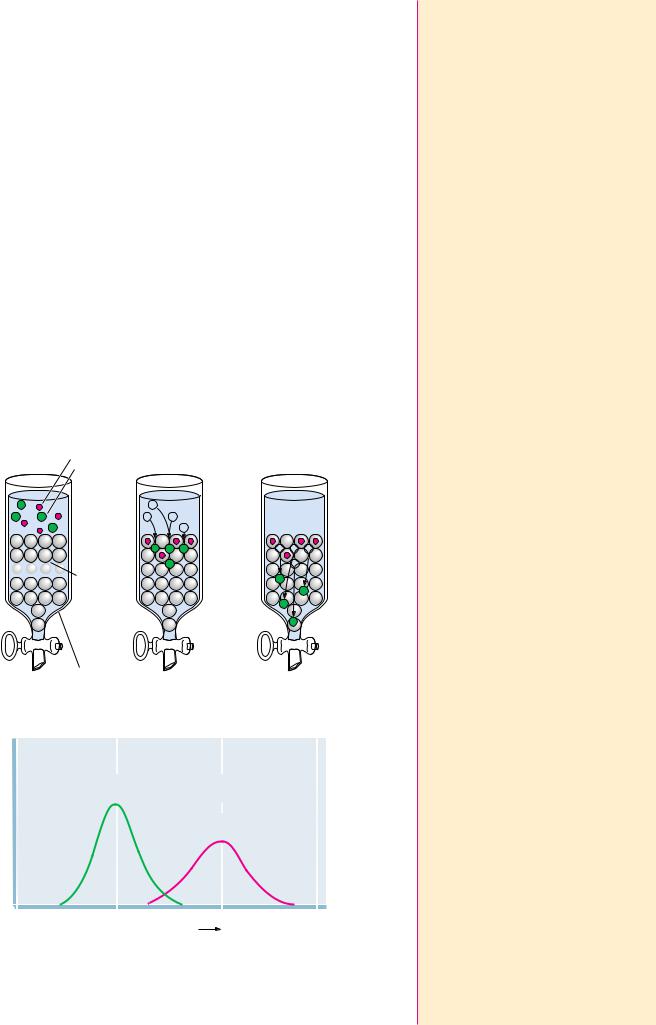
As a solution of molecules is passed through the column, the molecules passively distribute between Vo and Vi, depending on their ability to enter the pores (that is, their size). If a molecule is too large to enter at all, it is totally excluded from Vi and emerges first from the column at an elution volume, Ve, equal to Vo (Figure 5A.1). If a particular molecule can enter the pores in the gel, its distribution is given by the distribution coefficient, KD:
KD (Ve Vo)/Vi
where Ve is the molecule’s characteristic elution volume (Figure 5A.1). The chromatography run is complete when a volume of solvent equal to Vt has passed through the column.
Dialysis and Ultrafiltration
If a solution of protein is separated from a bathing solution by a semipermeable membrane, small molecules and ions can pass through the semipermeable membrane to equilibrate between the protein solution and the bathing solution, called the dialysis bath or dialysate (Figure 5A.2). This method is useful for removing small molecules from macromolecular solutions or for altering the composition of the protein-containing solution.
Ultrafiltration is an improvement on the dialysis principle. Filters having pore sizes over the range of biomolecular dimensions are used to filter solutions to select for molecules in a particular size range. Because the pore sizes in these filters are microscopic, high pressures are often required to force the solution through the filter. This technique is useful for concentrating dilute solutions of macromolecules. The concentrated protein can then be diluted into the solution of choice.
Electrophoresis
Electrophoretic techniques are based on the movement of ions in an electrical field. An ion of charge q experiences a force F given by F Eq/d, where E is the voltage (or electrical potential) and d is the distance between the electrodes. In a vacuum, F would cause the molecule to accelerate. In solution, the molecule experiences frictional drag, Ff, due to the solvent:
Ff 6 r v
where r is the radius of the charged molecule, is the viscosity of the solution, and v is the velocity at which the charged molecule is moving. So, the velocity of the charged molecule is proportional to its charge q and the voltage E, but inversely proportional to the viscosity of the medium and d, the distance between the electrodes.
Generally, electrophoresis is carried out not in free solution but in a porous support matrix such as polyacrylamide or agarose, which retards the movement of molecules according to their dimensions relative to the size of the pores in the matrix.
SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE)
SDS is sodium dodecylsulfate (sodium lauryl sulfate) (Figure 5A.3). The hydrophobic tail of dodecylsulfate interacts strongly with polypeptide chains. The number of SDS molecules bound by a polypeptide is proportional to the length (number of amino acid residues) of the polypeptide. Each dodecylsulfate contributes two negative charges. Collectively, these charges overwhelm any intrinsic charge that the protein might have. SDS is also a detergent that
|
|
|
|
|
|
|
Chapter 5 ● Appendix 155 |
|
|
O |
|
FIGURE 5A.3 ● The structure of sodium |
Na+ –O |
|
|
|
|
|
|
dodecylsulfate (SDS). |
|
S |
|
O |
CH2 |
CH2 CH2 CH2 CH2 CH3 |
|
|
|
|
|
|
|
|
CH2 |
CH2 CH2 CH2 CH2 CH2 |
|
|
O– |
|
|
|
|
Na+ |
|
|
disrupts protein folding (protein 3° structure). SDS-PAGE is usually run in the presence of sulfhydryl-reducing agents such as -mercaptoethanol so that any disulfide links between polypeptide chains are broken. The electrophoretic mobility of proteins upon SDS-PAGE is inversely proportional to the logarithm of the protein’s molecular weight (Figure 5A.4). SDS-PAGE is often used to determine the molecular weight of a protein.
Isoelectric Focusing
Isoelectric focusing is an electrophoretic technique for separating proteins according to their isoelectric points (pIs). A solution of ampholytes (amphoteric electrolytes) is first electrophoresed through a gel, usually contained in a small tube. The migration of these substances in an electric field establishes a pH gradient in the tube. Then a protein mixture is applied to the gel and electrophoresis is resumed. As the protein molecules move down the gel, they experience the pH gradient and migrate to a position corresponding to their respective pIs. At its pI, a protein has no net charge and thus moves no farther.
Two-Dimensional Gel Electrophoresis
This separation technique uses isoelectric focusing in one dimension and SDSPAGE in the second dimension to resolve protein mixtures. The proteins in a mixture are first separated according to pI by isoelectric focusing in a polyacrylamide gel in a tube. The gel is then removed and laid along the top of an SDS-PAGE slab, and the proteins are electrophoresed into the SDS polyacrylamide gel, where they are separated according to size (Figure 5A.5). The gel slab can then be stained to reveal the locations of the individual proteins. Using this powerful technique, researchers have the potential to visualize and construct catalogs of virtually all the proteins present in particular cell types. The ExPASy Molecular Biology World Wide Web server of the University of Geneva (located at URL: http://expasy.hcuge.ch/) provides access to a twodimensional polyacrylamide gel electrophoresis database named SWISS2DPAGE. This database contains information on proteins, identified as spots on two-dimensional electrophoresis gels, from many different cell and tissue types.
Relative electrophoretic  mobility
mobility
FIGURE 5A.4 ● A plot of the relative electrophoretic mobility of proteins in SDS-PAGE versus the log of the molecular weights of the individual polypeptides.
156 Chapter 5 ● Proteins: Their Biological Functions and Primary Structure
Isoelectric focusing gel
10
High
MW
4
electrophoresis of Direction
Low
MW
SDS-poly- |
Protein spot |
acrylamide |
|
slab |
|
FIGURE 5A.5 ● A two-dimensional electrophoresis separation. A mixture of macromolecules is first separated according to charge by isoelectric focusing in a tube gel. The gel containing separated molecules is then placed on top of an SDS-PAGE slab, and the molecules are electrophoresed into the SDS-PAGE gel, where they are separated according to size.
Affinity Chromatography
Recently, affinity purification strategies for proteins have been developed to exploit the biological function of the target protein. In most instances, proteins carry out their biological activity through binding or complex formation with specific small biomolecules, or ligands, as in the case of an enzyme binding its substrate. If this small molecule can be immobilized through covalent attachment to an insoluble matrix, such as a chromatographic medium like cellulose or polyacrylamide, then the protein of interest, in displaying affinity for its ligand, becomes bound and immobilized itself. It can then be removed from contaminating proteins in the mixture by simple means such as filtration and washing the matrix. Finally, the protein is dissociated or eluted from the matrix by the addition of high concentrations of the free ligand in solution. Figure 5A.6 depicts the protocol for such an affinity chromatography scheme. Because this method of purification relies on the biological specificity of the protein of interest, it is a very efficient procedure and proteins can be purified several thousandfold in a single step.
6.1 ● Forces Influencing Protein Structure |
159 |
the structure of the protein itself. The previous chapter described the details of primary protein structure. However, proteins do not normally exist as fully extended polypeptide chains but rather as compact, folded structures, and the function of a given protein is rarely if ever dependent only on the amino acid sequence. Instead, the ability of a particular protein to carry out its function in nature is normally determined by its overall three-dimensional shape or conformation. This native, folded structure of the protein is dictated by several factors: (1) interactions with solvent molecules (normally water), (2) the pH and ionic composition of the solvent, and most important, (3) the sequence of the protein. The first two of these effects are intuitively reasonable, but the third, the role of the amino acid sequence, may not be. In ways that are just now beginning to be understood, the primary structure facilitates the development of short-range interactions among adjacent parts of the sequence and also longrange interactions among distant parts of the sequence. Although the resulting overall structure of the complete protein molecule may at first look like a disorganized and random arrangement, it is in nearly all cases a delicate and sophisticated balance of numerous forces that combine to determine the protein’s unique conformation. This chapter considers the details of protein structure and the forces that maintain these structures.
6.1 ● Forces Influencing Protein Structure
Several different kinds of noncovalent interactions are of vital importance in protein structure. Hydrogen bonds, hydrophobic interactions, electrostatic bonds, and van der Waals forces are all noncovalent in nature, yet are extremely important influences on protein conformations. The stabilization free energies afforded by each of these interactions may be highly dependent on the local environment within the protein, but certain generalizations can still be made.
Hydrogen Bonds
Hydrogen bonds are generally made wherever possible within a given protein structure. In most protein structures that have been examined to date, component atoms of the peptide backbone tend to form hydrogen bonds with one another. Furthermore, side chains capable of forming H bonds are usually located on the protein surface and form such bonds primarily with the water solvent. Although each hydrogen bond may contribute an average of only about 12 kJ/mol in stabilization energy for the protein structure, the number of H- bonds formed in the typical protein is very large. For example, in -helices, the CPO and NOH groups of every residue participate in H bonds. The importance of H bonds in protein structure cannot be overstated.
Hydrophobic Interactions
Hydrophobic “bonds,” or, more accurately, interactions, form because nonpolar side chains of amino acids and other nonpolar solutes prefer to cluster in a
nonpolar environment rather than to intercalate in a polar solvent such as intercalate ● to insert between water. The forming of hydrophobic bonds minimizes the interaction of non-
polar residues with water and is therefore highly favorable. Such clustering is entropically driven. The side chains of the amino acids in the interior or core of the protein structure are almost exclusively hydrophobic. Polar amino acids are almost never found in the interior of a protein, but the protein surface may consist of both polar and nonpolar residues.
160 Chapter 6 ● Proteins: Secondary, Tertiary, and Quaternary Structure
FIGURE 6.1 ● An electrostatic interaction between the -amino group of a lysine and the-carboxyl group of a glutamate residue.
|
Main chain |
|
|
|
|
|
|
Main chain |
|
|
|
NH |
H2O |
Na |
+ |
|
|
Cl– |
HN |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
C |
|
|
|
|
|
O |
|
C |
|
O |
|
|
|
|
+ |
....... |
– |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HC |
CH2CH2CH2CH2NH3 |
O C CH2CH2 |
CH |
|
|
|
|
|
|
HN |
|
Lysine |
Cl– |
|
|
|
Glutamate |
|
NH |
|
|
|
C |
O |
|
|
|
|
|
O |
|
C |
|
|
|
|
|
Na+ |
|
|
H2O |
|
|
|
|
Electrostatic Interactions
Ionic interactions arise either as electrostatic attractions between opposite charges or repulsions between like charges. Chapter 4 discusses the ionization behavior of amino acids. Amino acid side chains can carry positive charges, as in the case of lysine, arginine, and histidine, or negative charges, as in aspartate and glutamate. In addition, the NH2-terminal and COOH-terminal residues of a protein or peptide chain usually exist in ionized states and carry positive or negative charges, respectively. All of these may experience electrostatic interactions in a protein structure. Charged residues are normally located on the protein surface, where they may interact optimally with the water solvent. It is energetically unfavorable for an ionized residue to be located in the hydrophobic core of the protein. Electrostatic interactions between charged groups on a protein surface are often complicated by the presence of salts in the solution. For example, the ability of a positively charged lysine to attract a nearby negative glutamate may be weakened by dissolved NaCl (Figure 6.1). The Na and Cl ions are highly mobile, compact units of charge, compared to the amino acid side chains, and thus compete effectively for charged sites on the protein. In this manner, electrostatic interactions among amino acid residues on protein surfaces may be damped out by high concentrations of salts. Nevertheless, these interactions are important for protein stability.
Van der Waals Interactions
Both attractive forces and repulsive forces are included in van der Waals interactions. The attractive forces are due primarily to instantaneous dipole-induced dipole interactions that arise because of fluctuations in the electron charge distributions of adjacent nonbonded atoms. Individual van der Waals interactions are weak ones (with stabilization energies of 4.0 to 1.2 kJ/mol), but many such interactions occur in a typical protein, and, by sheer force of numbers, they can represent a significant contribution to the stability of a protein. Peter Privalov and George Makhatadze have shown that, for pancreatic ribonuclease A, hen egg white lysozyme, horse heart cytochrome c, and sperm whale myoglobin, van der Waals interactions between tightly packed groups in the interior of the protein are a major contribution to protein stability.
6.2 ● Role of the Amino Acid Sequence in Protein Structure
It can be inferred from the first section of this chapter that many different forces work together in a delicate balance to determine the overall three-dimen- sional structure of a protein. These forces operate both within the protein structure itself and between the protein and the water solvent. How, then, does nature dictate the manner of protein folding to generate the three-dimensional



 Porous
Porous