

2. Анализ электронной структуры методами квантовой химии.
Квантово-химические методы исследования свойств органических молекул позволяют:
– получать информацию об электронной структуремолекулы, то есть распределении зарядов на атомах, характеристиках связей, а также
– вычислять энергию электронных уровней и устанавливать их конфигурацию, то есть заполнение электронами.
Методы квантовой химии объясняют фотофизические и спектральные свойстваорганических молекул, связывая эти свойства с особенностями строения, и поэтому широко используются для объяснения явлений цветности.
2.1. Способы описания энергетических состояний молекул.
Для качественного и количественного описания энергетического состояния молекул органическая химия использует два принципиально разных подхода. Это метод валентных связей, который возник из теории резонанса, иметод молекулярных орбиталей, имеющий квантово-механическое обоснование.
Метод валентных связей (ВС-метод)описывает строение органических молекул каксуперпозицию(гибрид, смесь) всех возможных классических и поляризованных структур.
Каждая из этих структур отличается:
расположением простых и кратных связей,
расположением зарядов и неподеленных пар электронов,
различной внутренней энергией.
Например, строение фенолав основном и возбужденном состояниях можно описать, по меньшей мере, пятью-резонансными структурами:
|
Состояния |
| ||||
|
Основное |
20% |
20% |
20% |
20% |
20% |
|
Возбужденное |
5% |
5% |
30% |
30% |
30% |

Однако основное и возбужденное состояние фенола может быть представлено различным вкладом (процентным содержанием) каждой их этих структур. Таким образом, описывается изменения в распределении электронной плотности, порядках связей и энергии молекулы при ее возбуждении светом.
Метод молекулярных орбиталей (МО-метод) представляет энергетические состояния молекулы как собственные значения энергии (Е) волновой функции () многоэлектронной системы (молекулы, иона, радикала), интерпретируя электроны как волноподобные частицы. Такое предложение (постулат) в аналитической форме имеет вид уравнения Шрёдингера.
(пси) Н = Е , гдеН - Гамильтониан или оператор энергии
Уравнение Шредингера в дифференциальной форме имеет точное решение только для одноэлектронных систем (молекулы водорода и гелия). Для нахождения функций многоэлектронных систем необходимо учитывать межэлектронные взаимодействия. Эта проблема решается в рамкахтеориисамосогласованного поля(ССП) с помощью итерационной процедуры. Для заданного набора одноэлектронных функций (орбиталей) орбиталей рассчитывается межэлектронное отталкивание, эта энергия затем используется для последующих расчетов новых наборов орбиталей и новых энергий отталкивания. Процесс продолжают до тех пор, пока не наступает сходимость и достигается самосогласование.
В связи с этим были предложены более совершенные операторы энергии, например, оператор Хартри-Фока(F). Его применяют в неэмпирических расчетах орбитальных энергий (ab initio) при решении системы уравнений Рутаана для различных органических молекул.
Широкому распространению квантовохимических методов способствовало применение приближенных методов расчета, которые отличаются большей простотой и наглядностью представления волновых функций. В этом случае делается ряд физически обоснованных предположений и упрощений.
Согласно теории МО многоэлектронная функция (пси)представляется какпроизведение волновых функций отдельных электронов, которые движутся (делокализованы) в поле всех атомных ядер ипод влиянием потенциала отталкивания остальных электронов системы.
(пси-функция)n
Эти одноэлектронныепси-функции (i) получили названиемолекулярных орбиталей (МО). Энергетическое состояние молекулы в целом оценивается как сумма энергий её молекулярных орбиталей.
![]()
Далее делается допущение, что молекулярные орбитали iформируются каклинейная комбинация атомных орбиталей (АО) – одноэлектронных волновых функций атомовn (фи). При этом волноваяпси-функция выражается как суммаиндивидуальных атомных орбиталей, умноженных на соответствующие весовые коэффициенты.
![]()
В методе ЛКАО отдельные атомы рассматриваются явно. Физическое обоснование этого приближения основано на том, что эффективная энергия электрона в момент его нахождения вблизи одного из атомных ядер молекулы в основном определяется потенциалом этого ядра.
Атомные орбиталиn , которые включены в молекулярную орбитальi,называются базисными орбиталями. Минимальный базисный набор для молекул, содержащих атомыH,C,N,O, будет состоять из 1s, 2s, 2px,y,zорбиталей. Выражение ЛКАО (линейная комбинация атомных орбиталей) предполагает, что АО (n) принадлежат различным атомам молекулы, причем каждыйатом предоставляет для формирования МО только определенную АО.
Молекулярные орбитали (пси) iобладают всеми свойствами волновых функций.
1.Квадраты коэффициентовc2при АО оценивают вероятность нахождения (пребывания) электрона на данной АО, то есть, представляет долю электронной плотности на данной АО.
2. Численные значения коэффициентов при АО таковы, что сумма их квадратов равна 1 и соответствует заряду электрона. Тогда для любой МО можно записатьусловие нормировки:
(пси) ic21i + c22i + ... + c2ni = 1
nАО формируетn молекулярных орбиталей:

фи-Функция (n) не имеет физического смысла, но её решение позволяет описать форму электронных облаков канонических АО. В отличие от собственной функции её квадрат представляет плотность вероятности нахождения электрона в определенной области пространства, то есть информирует о распределении электронной плотности в области ковалентного связывания.
Приближенный квантово-химический расчет для сложных молекул становятся возможными благодаря модификации способа решения уравнения Шредингера. В рамках этого уравнения вариационный метод сводит квантовомеханическую задачу нахождения значений собственных энергий Eiи коэффициентовciволновых функцийi(МО) к математической задаче решения системы линейных однородных уравнений относительно значений коэффициентаci, имеющей вид:

Решению системы уравнений предшествует оценка значенийоператоров энергии(H) иинтегралов перекрывания(S). Указанные интегралы рассчитывают на основе соответствующих атомных функций или заменяют параметрами.
В расчетном методе МО Хюккеля используют три эмпирических параметра.
–Кулоновский интеграл, который определяют из выражения:
![]() (интеграл
оператора энергии,=)
(интеграл
оператора энергии,=)
Кулоновский интеграл учитывает энергию электростатического взаимодействия i-электрона с ядром, то есть является мерой электроотрицательности данного атома. Заметим, что электроотрицательность элемента рассматривается, как способность атома удерживать свои электроны или мера его сродства к электрону. В пределах одного периода электроотрицательность возрастает с увеличением заряда ядра.
–Резонансный интеграл, который определяется из выражения:
![]() (интеграл
оператора энергии,)
(интеграл
оператора энергии,)
Резонансный интеграл учитывает энергию электрона, находящегося в поле соседних атомов и, которые образуют химическую связь, то есть служитмерой энергии связи между атомами. В методах МОХявляется условной (относительной) единицей энергии.
S – Интеграл перекрывания, который определяется из выражения:
![]()
Интеграл перекрывания учитывает степень перекрывания (смешивания) АО атомови, которые образуют химическую связь. Эффективность смешивания соотноситсяс геометрическими условиями перекрывания АО, что, в конечном счете, определяет энергию формирующейся молекулярной орбитали.
В простом методе МО Хюккеля Sпринимается равным нулю, но в расширенном методе Хюккеля (РМХ), многих полуэмпирических методах, а также методе возмущения молекулярных орбиталей может принимать значения от 0 до 1.
Используя эти параметры, преобразуют такую систему уравнений в систему вида:
![]()
Систему уравнений решают методом матричной алгебры и в итоге получают:
nзначенийсобственной энергиимолекулярных орбиталейEi(по числу АО, формирующих МО) и
n наборовсобственных коэффициентов сi, соответственно числу сформированных молекулярных орбиталей.
В методах МОХ энергия МО записывается в единицах в виде выражения:
E = y, гдеу- численный коэффициент
в других полуэмпирических методах в единицах энергии - эВ.
Например, решение квантово-механической задачи для бензола с учетом в базисе только шести p-орбиталей (-приближение) дает6 значенийсобственной энергии Е1-Е6 и по 6 наборовкоэффициентовciдля каждой из шести МО(см. рис).
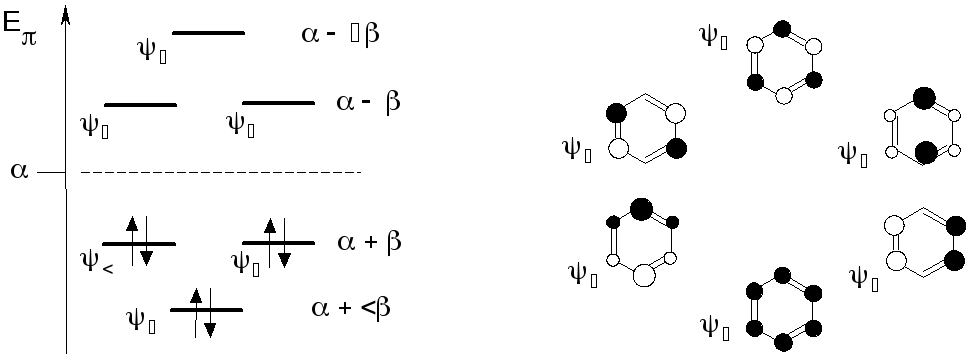

Молекулярные орбитали заселяютсятакже как и АО, в соответствии с правилом Паули, для нихчисло заполненияNравно 0,1 и 2.
Шесть -электронов бензола заселяют попарно три меньшие по энергии МО. Орбиталиоказываютсязанятыми МО. Согласно расчетуиимеют одинаковую энергию, такие орбитали называютсявырожденными. Среди занятых эти МО наибольшие по энергии и называютсявысшими занятыми МО (ВЗМО).
Симметрично занятым в бензоле сформированы три свободные иливакантныемолекулярные-орбитали4, и6. Две из них также являются вырожденными МО. Среди вакантных МО орбитали4,меньше по энергии и поэтому называютсянизшими вакантными МО (НВМО).
Наиболее близкие по энергии ВЗМО и НВМО принято называть граничными орбиталями.
Собственные коэффициентыciМО являются основой для оценки некоторых физических и химических свойств молекулы. Так как коэффициенты находят из условия нормировки, каждое значениеciпозволяет оценить вклад каждой АО в формируемуюi-МО. Еслиciравно 1, то данная МО полностью локализована на-й АО. Такая МО называетсянесвязывающей молекулярной орбитальюи обозначается как n-МО. В молекулах, которые содержат гетероатомы (азот, кислород, сера и др.), неподеленные пары электронов гетероатомов могут занимать несвязывающие МО.
Значения собственных коэффициентов МО позволяют вычислить основные параметры электронной структуры молекулы. Этими параметрами являются:
электронная плотность на данной АО для соответствующей МО оценивается квадратом коэффициента c2i с учетом заполнения МО электронами;
![]()
полная электронная плотностьна атомеподсчитывается суммированием электронной плотности по всем МО, принадлежащим к какому-либо атому;
![]()
электронная плотность в области перекрывания иАОдля соответствующейi‑МО оценивается произведением коэффициентовcici с учетом заполнения МО электронами;
![]()
порядок связи между соседними атомами можно вычислить суммирование межорбитальной электронной плотности по всем МО.
![]()
Порядок связи rв молекулы находится в соответствии с ее длиной, дипольным моментом и силовой постоянной связи.
Далее отметим, поскольку суммирование электронных плотностей q иr проводят с учетом заселенности МО электронами, тогдавклад в связывание между атомамив молекуледелают только занятые МО, поэтому их называютсвязывающими МО. Напротив, свободные МО называютразрыхляющими МО, поэтому переход электрона на эти МО при фотовозбуждении дестабилизирует молекулярную структуру.
Т аким
образом, методы МО устанавливают
наглядную и логичную связь между
строением молекулы и её электронной
структурой, энергией и природой
молекулярных орбиталей (МО).
аким
образом, методы МО устанавливают
наглядную и логичную связь между
строением молекулы и её электронной
структурой, энергией и природой
молекулярных орбиталей (МО).
Положение полосы поглощения в электронном спектре и, следовательно, наличие или отсутствие окраски у вещества,
– можно связать с конкретным электронным переходом с занятой МО на вакантную МО (более высокий энергетический уровень),
– а затем вычислить энергию электронного перехода и сопоставить с энергией реально поглощаемых фотонов света.
Таким образом, отпадает необходимость объяснять окраску соединения превращением и резонансом классических и поляризованных структур, как в случае метода валентных связей.