

Желчные кислоты
ЖЕЛЧНЫЕ КИСЛОТЫ, монокарбоновые гидроксикислоты, относящиеся к классу стероидов. Почти все желчные кислоты - производные прир. холановой к-ты, синтезируемой из холестерина в печени.
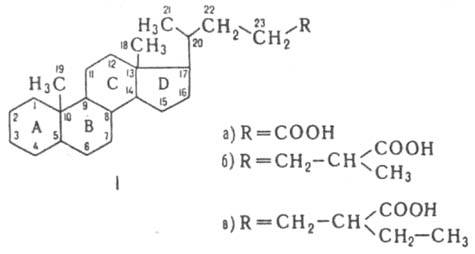
Сначала синтезируются две первичные желчные кты – холевая и хенодезоксихолевая. Потом вторичные: дезоксихолевая и литохолевая. Функционируют они в виде конъюгантов с глицином или таурином. Конъюганты в печени образуются в две стадии:
R-COOH+ATP+KoASH = R – CO~SKoA+AMP+PP
R-CO~SKoA+H2N-CH2-COOH(глицин) = R-CO-NH-CH2-COOH+KoASH
Или
R-CO~SKoA+ H2N-CH2-CH2-SO3H(таурин) = R-CO-NH-CH2-CH2-SO3H+KoASH
Желчные кислоты эмульгируют жиры.
Стероидные гормоны
Общим предшественником стероидных гормонов является холестерин.
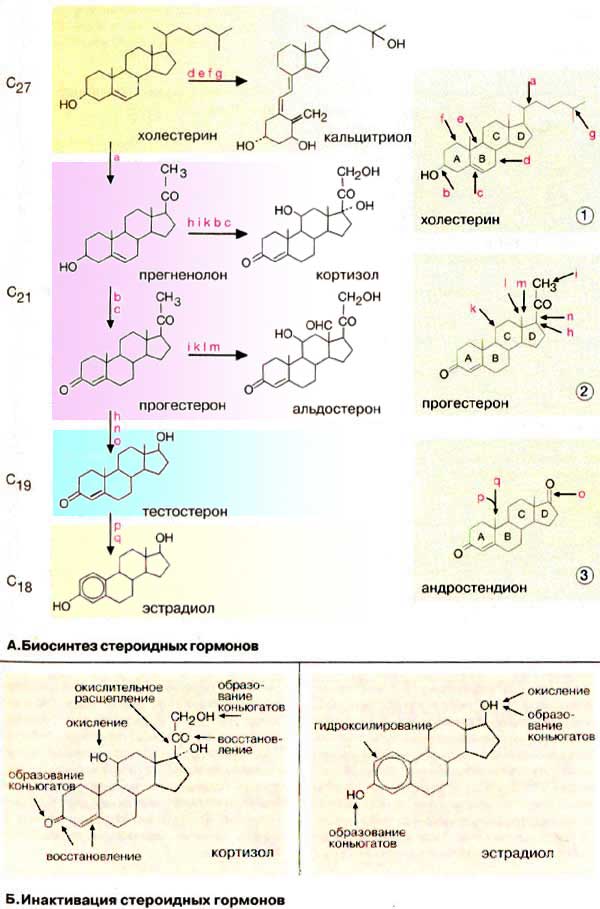
Путь биосинтеза. Биосинтез каждого гормона состоит из множества последовательных ферментативных реакций. В качестве примера рассмотрим биосинтез прогестерона (А) см. рис. 397). Биосинтез начинается с расщепления боковой цепи холестерина между С-20 и С-22 (а). Стероидное соединение с укороченной боковой цепью носит название прегненолон. Последующие стадии, окисление гидроксигруппы при С-3 (b) и сдвиг двойной связи от С-5 к С-4 (с) приводят к образованию прогестерона.
Приведенные на схеме стероиды объединены в подгруппы по числу углеродных атомов. Холестерин и кальцитриол являются С27-стероидами. Соединения с укороченной на 6 атомов углерода боковой цепью, прогестерон, кортизол и альдостерон, составляют группу С21-стероидов. В ходе биосинтеза тестостерон полностью утрачивает боковую цепь и поэтому его относят к С19-стероидам. При биосинтезе эстрадиола на стадии образования ароматического цикла теряется ангулярная метильная группа и, следовательно, эстрадиол является С18-стероидом.
В процессе биосинтеза кальцитриол подвергается фотохимической реакции раскрытия кольца В. Поэтому его относят к «секостероидам». Однако по своим биохимическим свойствам он является типичным стероидным гормоном.
2.Специализированные пути метаболизма цикл. А,к- фенилаланина и тирозина.. Заболевания, связанные с нарушением обмена фенилаланина и тирозина.
Фенилаланин относится к незаменимым аминокислотам, поскольку ткани животных не обладают способностью синтезировать его бензольное кольцо. В то же время тирозин полностью заменим при достаточном поступлении фенилаланина с пищей. Объясняется это тем, что основной путь превращения фенилаланина начинается с его окисления (точнее, гидрокси-лирования) в тирозин (рис. 12.6). Реакция гидроксилирования катализируется специфической фенилаланин-4-монооксигеназой, которая в качестве кофермента содержит, как все другие гидроксилазы, тетрагидро-биоптерин. Блокирование этой реакции, наблюдаемое при нарушении синтеза фенилаланин-4-монооксигеназы в печени, приводит к развитию тяжелой наследственной болезни – фенилкетонурии (фенилпировиноградная олигофрения). В процессе трансаминирования тирозин превращается в n-оксифенилпировиноградную кислоту, которая под действием специфической оксидазы подвергается окислению, декарбоксилированию, гидро-ксилированию и внутримолекулярному перемещению боковой цепи с образованием гомогентизиновой кислоты; эта реакция требует присутствия аскорбиновой кислоты, роль которой пока не выяснена. Дальнейшее превращении егомогентизиновой кислоты в малеилацетоуксусную кислоту катализируется оксидазой гомогентизиновой кислоты. Малеилацетоуксус-ная кислота под действием специфической изомеразы в присутствии глу-татиона превращается в фумарилацетоуксусную кислоту, подвергающуюся гидролизу с образованием фумаровой и ацетоуксусной кислот. Фенилаланин и тирозин являются также предшественниками меланинов. В этом важном биологическом процессе, обеспечивающем пигментацию кожи, глаз, волос, активное участие принимает фермент тирозиназа.
Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллин-га) — одно из наиболее частых наследственных заболеваний, чаще обусловлено недостаточностью фенилаланин гидроксилазы (261600, 12q24.1); при отсутствии своевременного лечения приводит к тяжёлой умственной отсталости. Тип наследования аутосомно-рецессивный. Больные гомозиготны по гену фенилкетонурии, а их родители гетерозиготны.
Вследствие дефекта гена фенилаланин гидроксилазы (ФАГ-ген) развивается недостаточность фермента, и как следствие наступает блок в нормальном превращении фенилаланина в аминокислоту тирозин . Фенилаланин накапливается в организме и его концентрация в крови повышается в 10-100 раз. Далее он превращается в фенилпировиноградную кислоту, оказывающую токсическое влияние на нервную систему. Накопление фенилаланина в организме происходит постепенно, поэтому клиническая картина развивается медленно. В связи с этим очень важна ранняя диагностика фенилкетонурии
Алкаптонурия возникает вследствие мутации гена, кодирующего синтез оксидазы гомогентезиновой кислоты. Данная патология характеризуется аутосомно-рецессивным типом наследования. Алкаптонурией чаще болеют мужчины. Ген оксидазы гомогетинзиновой кислоты человека (HGD) локализован на длинном плече 3 хромосомы человека . В нормальных условиях гомогентезиновая кислота — промежуточный продукт распада тирозина и фенилаланина — переводится в малеилацетоуксусную кислоту, из которой в конечном счёте образуются фумаровая и ацетоуксусная кислоты, вступающие в другие биохимические циклы. Из-за дефекта фермента этот процесс тормозится, и остающаяся в избытке гомогентезиновая кислота превращается полифенолоксидазой в хиноновый полифенол (алкаптон или бензохинонацетат), который и выводится почками. Не полностью экскретируемый мочой алкаптон откладывается в хрящевой и другой соединительной ткани, обусловливая их потемнение и повышенную хрупкость. Чаще всего вперёд появляется пигментация склер и ушных хрящей.
.