

Глава 3. Функциональные производные алканов
В этой главе рассматриваются производные алканов с одной или несколькими гетерофункциональными группами X. Общая формула таких монофункциональных производных R–X, где группировка X представлена гетероатомом или их совокупностью. Это атомы галогенов (-F, -Cl, -Br, -I), гидроксильная и алкоксильная группы (-OH, -OR), меркапто- и алкилтиогруппы (-SH, -SR), аминогруппы (в т.ч. замещенные: -NH2, -NHR, -NR2), нитрогруппа (-NO2) и др. Таким образом, общий структурный фрагмент, присутствующий в этих соединениях, можно представить в виде:
 ,
,
где Х — перечисленные функциональные группы -F, -Cl, -Br, -I, -OH, -OR, -SH, -SR, -NH2, -NHR, -NR2, -NO2.
3.1. ОбщноСть Химических свойств
Молекулы, содержащие фрагмент С–Х, в которых атом углерода связан с атомами галогена, кислорода, серы или азота, объединяет то, что это прежде всего электроотрицательные атомы, а значит, связь С–Х полярна, причём во многих случаях — сильно полярна. В отсутствие -связей в углеводородной основе гетероатом функциональной группы Х может взаимодействовать с соседним атомом углерода только посредством индуктивного эффекта, а так как гетероатом функциональной группы обладает бóльшей электроотрицательностью, чем углерод, то индуктивный эффект функциональной группы Х всегда акцепторный. При этом на соседнем атоме углерода наблюдается дефицит электронной плотности (возникает частичный положительный заряд):
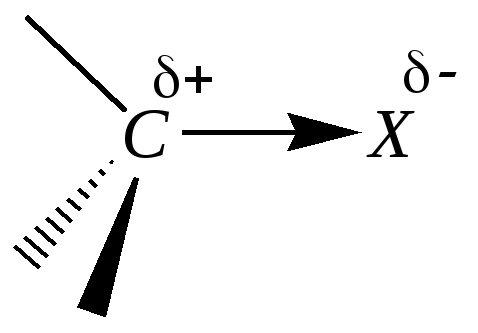
Наличие в молекулах электрофильного центра (С+) создаёт условия для нуклеофильной атаки по этому атому углерода с возможным разрывом -связи С–Х и образованием новой -связи между углеродом и нуклеофильной частицей. Таким образом, в результате возможно замещение одних атомов акцепторов или атомных группировок (Х) на другие. Это свидетельствует о том, что для таких соединений должны быть характерны реакции нуклеофильного замещения (SN).
Кроме того, электроноакцепторный эффект, передаваясь по цепи углеродных атомов, поляризует и С–Н-связи. В связи с этим С–Н-связь соседнего (-углеродного) атома становится значительно более полярна:
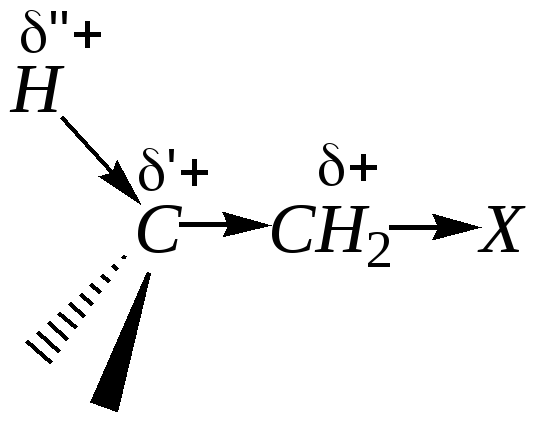
Этот атом водорода также может быть подвергнут атаке основания, что приводит к протеканию реакций отщепления (Е).
Рассмотрим более подробно реакции нуклеофильного замещения и реакции отщепления.
3.1.1. Реакции нуклеофильного замещения
Наиболее активны в реакциях нуклеофильного замещения должны быть галогеналканы RF, RCl, RBr и RI, так как в их молекулах при замещении образуется стабильный анион уходящей группы Х¯, представляющий собой один из галогенид-ионов, то есть анион сильной кислоты. Это подтверждается многочисленными примерами замещения атомов галогена в галогеналканах, например, на гидрокси-, алкокси-, амино-, циано-, нитрогруппу. Напротив, амины должны обладать наименьшей реакционной способностью, так как аммиак и амины — очень слабые кислоты и, соответственно, сопряжённые им основания, то есть анионы ¯NH2, ¯NHR, ¯NR2, высокореакционноспособны и потому неустойчивы (легко присоединяют протон). Гидроксильная группа в спиртах также может замещаться в реакциях со многими нуклеофилами, однако в более жёстких условиях. Ещё труднее замещается алкоксильная группа. Гидроксильная и алкоксильная группы замещаются только в кислой среде, в которой уходящей частицей является не анион, а молекула (соответственно воды или спирта). Аминогруппа достаточно устойчива к замещению, случаи её нуклеофильного замещения редки, реакции протекают в очень жёстких условиях и только для аммониевых солей. В связи с этим наиболее широк спектр SN-реакций галогеналканов (гл. 3.2).
Реакции нуклеофильного замещения у sp3-гибридизованного атома углерода наиболее изучены в органической химии. Так же, как и в случае радикального замещения, здесь предполагается разрыв s-связи в молекуле исходного вещества, называемом ещё субстратом, и образование новой s-связи в продукте реакции. Однако нуклеофильное замещение относится к реакциям ионного типа, поэтому молекула исходного вещества (RX) должна быть поляризована, причём у заместителя X должна быть достаточно высокая эффективная электроотрицательность. Общая схема реакции может быть представлена в следующем виде:
![]()
Атакующий агент Y, называемый нуклеофилом, за счёт неподелённой пары электронов атакует в молекуле субстрата положительно заряженный центр. Реакция сопровождается гетеролитическим разрывом s-связи в молекуле субстрата, заместитель X уходит вместе с парой электронов. Новая ковалентная связь образуется за счёт пары нуклеофильного реагента координационным путем.
Нуклеофильными реагентами могут быть самые разнообразные частицы, но они должны обязательно иметь неподелённую электронную пару. Это, например, анионы HО¯, RO¯, ¯NH2, F¯, Cl¯, Br¯, I¯, CN¯, H¯, ¯CH2-R и нейтральные молекулы H2O, ROH, NH3, RNH2, RRNH, H2S, RSH. Нуклеофильными свойствами обладают и такие соединения, как непредельные и ароматические углеводороды.
Субстратами могут быть полярные молекулы, имеющие атом углерода с эффективным положительным зарядом и замещаемую группу X. Атом углерода здесь называется электрофильным центром. Группа X, называемая также уходящей группой, или нуклеофугом, обладает высокой электроотрицательностью и может уходить как в виде аниона, так и в виде незаряженной молекулы.
В реакциях нуклеофильного замещения в зависимости от природы субстрата, нуклеофила, уходящей группы и от условий реакции могут реализоваться несколько различных механизмов. Для таких реакций наиболее распространёнными являются механизм бимолекулярного нуклеофильного замещения, обозначаемый SN2, и мономолекулярного нуклеофильного замещения, обозначаемый SN1.
Механизм бимолекулярного нуклеофильного замещения
Реакция бимолекулярна, потому что протекает при столкновении двух частиц: нуклеофила и молекулы субстрата. Скорость реакции при этом зависит от концентрации субстрата и от концентрации атакующих нуклеофильных частиц. Нуклеофил атакует положительно заряженный центр молекулы субстрата с электростатически более выгодной «тыльной» стороны, так как в этом случае на него не действует одноимённый заряд нуклеофуга. Реакция представляет собой одностадийный процесс. Связь C–Y образуется одновременно с разрывом C–X связи.
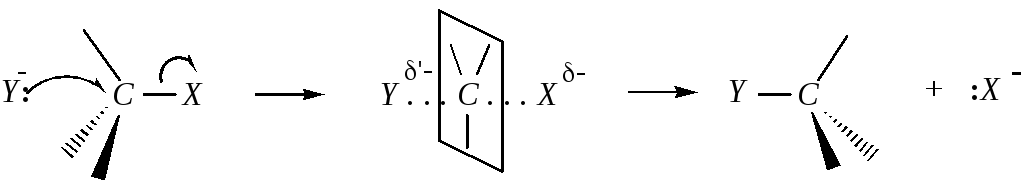
Энергия, необходимая для разрыва C–X-связи, поставляется за счёт синхронного процесса образования связи C–Y. Как только группа Y включается в переходное состояние, группа Х должна уйти, поскольку атом углерода не может иметь более восьми электронов на внешнем уровне. В переходном состоянии исходная sp3-гибридизация атома углерода изменяется на sp2-гибридизацию с примерно перпендикулярной р-орбиталью. В переходном состоянии нуклеофильный реагент, центральный атом углерода и нуклеофуг находятся на прямой линии, поэтому, если подход нуклеофила со стороны, противоположной уходящей группе, невозможен, например, вследствие особенностей строения субстрата, бимолекулярная реакция также становится невозможной. Три нереагирующие группы субстрата и центральный атом углерода примерно компланарны, то есть находятся в одной плоскости. Они будут строго компланарны, если входящая и уходящая группы одинаковы. В других случаях возможно как более раннее переходное состояние (связи центрального атома углерода ещё не приняли тригональную конфигурацию, порядок связи С...Х больше порядка связи C...Y), так и более позднее.
Стереохимию процесса бимолекулярного нуклеофильного замещения легко наблюдать на примере гидролиза оптически активного субстрата. Три нереагирующие группы при атаке как бы «выворачиваются» внутрь. Поэтому иногда говорят и про атом углерода, что он «выворачивается», но чаще всего пользуются термином «обращение конфигурации» атома углерода, имея в виду изменение пространственного расположения присоединённых к нему групп. Действительно, если бы группы Х и Y имели одинаковую химическую природу (например, в реакции изотопного обмена при замещении 35Сl на 37Сl), то оказалось бы, что продукт реакции вращает луч плоскополяризованного света в противоположном направлении по сравнению с исходным веществом и является его зеркальным отражением. Этот процесс сравнивают с выворачиванием зонтика на ветру. Такое изменение конфигурации известно ещё под названием «вальденовское обращение». Все реакции бимолекулярного нуклеофильного замещения сопровождаются вальденовским обращением независимо от строения субстрата.
Механизм мономолекулярного замещения
Идеальный вариант механизма SN1 включает две стадии:
1)
![]()
2)
![]()
Первая стадия — это медленная ионизация субстрата, и именно она определяет скорость реакции. Диссоциации молекул на свободные ионы предшествует переходное состояние, в котором происходит увеличение длины связи С–Х и постепенное перемещение электронной пары к уходящей группе. Затем образуется ионная пара. Распад её на ионы происходит почти всегда при участии молекул полярного растворителя. Практически механизм SN1 осуществляется легко только в полярных растворителях. Вторая стадия — это быстрое взаимодействие промежуточного карбокатиона с нуклеофилом.
Таким образом, скорость всей реакции в целом зависит только от скорости наиболее медленной первой стадии, в которой принимают участие только молекулы субстрата. Поэтому реакция мономолекулярна, а её скорость зависит только от концентрации исходного субстрата.
Образующаяся в результате протекания процесса замещения частица Х¯ может снижать скорость реакции из-за её обратимости. Поэтому во многих случаях можно добавками солей, содержащих анионы Х¯, замедлить реакцию. Такое понижение скорости реакции, вызванное добавлением Х¯, называется эффектом общего иона.
В общем случае для SN1-реакций скорость не должна зависеть от природы нуклеофила и его концентрации.
Стереохимия мономолекулярного нуклеофильного замещения менее однозначна, чем замещения SN2-типа. В идеальном случае если процесс включает образование свободного карбокатиона, то последний должен быть планарен, то есть иметь плоскую конфигурацию, соответствующую sp2-гибридизации орбиталей. Нуклеофил должен с одинаковой скоростью атаковать карбокатион с обеих сторон плоскости, что приведёт к образованию двух новых молекул субстрата, являющихся энантиомерами по отношению друг к другу. В результате образуется рацемическая смесь.
Для многих реакций довольно легко утверждать, что при данных условиях они следуют по механизму либо SN1, либо SN2. Однако в некоторых случаях механизм реакции охарактеризовать значительно труднее. Существуют промежуточные случаи, так называемая пограничная область механизмов, то есть механизм реакции не является ни «чистым» SN1, ни «чистым» SN2, а относится к промежуточному типу. Это можно представить следующей схемой:
![]()
I II III IV V,
где II — тесная ионная пара, III — рыхлая ионная пара, IV и V — диссоциированные ионы, каждый из которых окружён молекулами растворителя.
Таким образом, SN1- и SN2-реакции могут быть объяснены ион-парным механизмом. Субстрат диссоциирует с образованием промежуточной ионной пары, которая затем превращается в продукты. Различие между механизмами SN1 и SN2 заключается в том, что в первом случае нуклеофильной атаке подвергаются диссоциированные ионы (IV), а во втором случае нуклеофил атакует системы I, II и, возможно, III. Так как в общем случае нуклеофильной атаке субстрат может подвергаться на любой стадии превращения по приведённой схеме, то чаще всего можно лишь утверждать, что тот или иной механизм близок к SN1 или SN2.
Факторы, влияющие на механизм и скорость нуклеофильного замещения
Одни и те же факторы могут совершенно различным образом влиять на скорость реакций, протекающих по «чистому» SN1- или SN2-механизму. Поэтому какие-то из них способствуют протеканию реакций по мономолекулярному механизму, а какие-то — по бимолекулярному.
1. Влияние структуры субстрата. Увеличение пространственного объёма заместителей при реакционном центре субстрата уменьшает скорость бимолекулярного замещения, так как реакционный центр становится менее доступным атаке нуклеофила. При этом при переходе от бромметана к бромэтану скорость SN2-реакции уменьшается в 145 раз, а к 2-бромпропану — в 18 000 раз.
![]()
бромметан бромэтан 2-бромпропан
Однако скорость SN1-реакции в этом ряду будет возрастать, так как влияние электронных эффектов заместителей в субстрате в большинстве случаев значительно сильнее проявляется в реакциях мономолекулярного замещения. Поэтому очевидно, что при переходе от первичных систем к вторичным и третичным скорость по этому механизму должна увеличиваться. Это можно объяснить увеличением стабильности алкильных катионов:
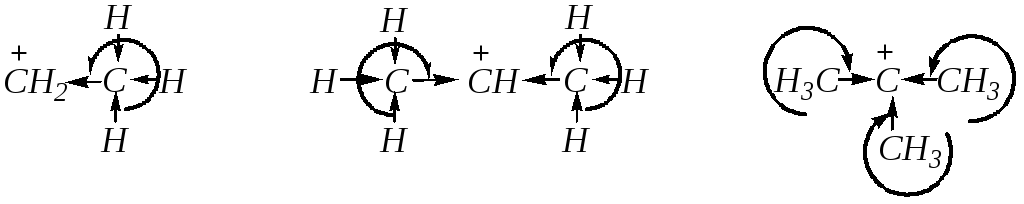 ,
,
зависящей, в частности, от количества метильных групп вокруг положительно заряженного атома углерода, которые обладают электронодонорным индуктивным эффектом и поэтому компенсируют заряд реакционного центра. В этом же ряду возрастает и величина эффекта сверхсопряжения р-орбитали карбокатионного атома углерода с электронами С–Н-связей. Поэтому высокая скорость реакций нуклеофильного замещения может быть характерна и для первичных, и для третичных алкилгалогенидов. В первом случае — за счёт лёгкости взаимодействия по SN2-механизму (свободный доступ реакционного центра, нет стерических препятствий), во втором — по SN1-механизму (лёгкость диссоциации субстратов, стабильность образующегося карбокатиона). Вторичные алкилгалогениды в большинстве случаев должны реагировать по смешанному механизму, причём скорость реакций у них будет относительно небольшой, так как есть препятствия для протекания и мономолекулярного, и бимолекулярного замещения.
Введение заместителей в молекулу субстрата по-разному будет влиять на скорость мономолекулярной и бимолекулярной реакций. Электронодонорные заместители должны ещё более стабилизировать образующийся катион и, следовательно, увеличивать скорость мономолекулярного замещения.
Влияние электронных эффектов заместителей на скорость бимолекулярной реакции не столь однозначно. Но большинство SN2-реакций ускоряется электроноакцепторными заместителями — в этих случаях скорость реакции определяется лёгкостью взаимодействия нуклеофильной частицы с положительно заряженным реакционным центром субстрата. В других случаях скорость бимолекулярной реакции зависит от способности нуклеофугной группы отщепляться от реакционного центра, и влияние природы заместителей будет противоположным — таким же, как и в мономолекулярных реакциях.
2. Влияние природы нуклеофила. В реакциях нуклеофильного замещения практически любая нейтральная или отрицательно заряженная частица, имеющая неподелённую пару электронов, может быть нуклеофилом. Скорости мономолекулярных реакций не зависят от природы нуклеофила, так как он не принимает участия в лимитирующей стадии, поэтому нуклеофильность реагента, то есть способность предоставлять электронную пару для образования ковалентной связи при взаимодействии с положительно заряженным центром в субстрате, влияет только на скорость SN2-реакций. Для этих реакций, протекающих в растворе, можно отметить несколько основных принципов, которые определяют влияние нуклеофила на скорость.
Во-первых, нуклеофильность аниона всегда выше, чем у соответствующей нейтральной молекулы. Так, ¯OH сильнее, чем H2O; ¯NH2 сильнее, чем NH3, и т.д.
Во-вторых, при сравнении нуклеофилов, атакующие атомы которых находятся в одном периоде периодической системы, порядок нуклеофильности приблизительно совпадает с порядком основности. Поэтому
R3C¯>R2N¯>RO¯>F¯
Электронодонорные заместители увеличивают нуклеофильность реагента, так, у RO¯ нуклеофильность выше, чем у HO¯; у RSH — чем у H2S, и т.д. Высокой нуклеофильностью обладают неустойчивые анионы, в частности карбанионы, так как образование аниона для атома углерода энергетически невыгодно (из-за его низкой электроотрицательности), и потому такие частицы обладают большой потенциальной энергией.
В-третьих, нуклеофильность возрастает сверху вниз в группах периодической системы (с увеличением радиуса атома), хотя основность в этом ряду падает. Так, обычный порядок нуклеофильности галогенидов выглядит следующим образом: I¯>Br¯>Cl¯>F¯. Аналогично любой серосодержащий нуклеофил сильнее соответствующего кислородсодержащего аналога, и то же справедливо для соединений, содержащих фосфор и азот. Это объясняется лёгкостью поляризации атомов и ионов большего размера и меньшей энергией сольватации этих ионов.
В-четвёртых, чем свободнее нуклеофилы, тем больше скорость; сольватация снижает скорость реакции. Так, протонные растворители (см. ниже) снижают нуклеофильную силу реагента за счёт образования водородных связей, если нуклеофильным центром является сильно электроотрицательный элемент (F, O, N).
Однако эти правила не могут учесть всех факторов, влияющих на нуклеофильность реагента. Так, часто определённую роль играют стерические препятствия. Например, трет-бутилат-ион (CH3)3CO¯ — более сильное основание, чем HO¯ или C2H5O¯, но значительно менее сильный нуклеофил, так как его большой пространственный объём затрудняет близкий подход к субстрату.
Таким образом, активность наиболее распространённых нуклеофилов уменьшается в ряду (для SN2-реакций в протонных растворителях):
RS¯>C2H5S¯>I¯>CN¯>HO¯>Br¯>C2H5O¯>Cl¯>CH3COO¯>H2O
3. Влияние растворителей и катализаторов. В мономолекулярном замещении на первой стадии из нейтральной молекулы субстрата образуются ионы, они легко сольватируются молекулами полярных растворителей, особенно протонных. Поэтому протонные высокополярные растворители будут способствовать протеканию реакции по механизму SN1.
В лимитирующей стадии бимолекулярного замещения принимает участие нуклеофильная частица. Поэтому применение протонного растворителя приведёт к её дезактивации за счёт образования водородных связей с атомами водорода растворителя и замедлению реакции. В апротонных растворителях нуклеофильные реагенты сохраняют высокую реакционную способность. Кроме того, сольватируя катионы, апротонные растворители способствуют диссоциации молекул реагента на ионы и, следовательно, увеличивают нуклеофильную силу реагента. Таким образом, полярные апротонные растворители способствуют SN2-реакции. Поэтому кислая среда, как правило, не способствует протеканию бимолекулярных реакций, и для этих реакций предпочтительна нейтральная или щелочная среда, так как сильные нуклеофилы — это обычно сильные основания.
В качестве катализаторов в реакциях нуклеофильного замещения используются кислоты Льюиса, то есть галогениды бора, алюминия, железа, цинка, кадмия, ртути, меди и другие. Эти вещества способны акцептировать анионы из раствора за счёт валентных орбиталей металла, и их применение может лишь замедлить процесс бимолекулярного замещения, но способствует протеканию SN1-процесса, так как облегчает диссоциацию субстрата, при этом катализатор не взаимодействует с образующимися карбокатионами субстрата.
4. Влияние природы уходящей группы. В ходе реакции уходящая группа отщепляется вместе с парой электронов.
В реакциях, осуществляемых по механизму мономолекулярного замещения, чем легче будет уходящая группа отщепляться, тем быстрее пойдёт реакция, так как именно разрыв связи С–Х и является лимитирующей стадией реакции, осуществляемой по механизму SN1. На лёгкость же отщепления влияет не только энергия диссоциации связи, но и стабильность нуклеофугной группы как свободной частицы. Так, при отрыве галогенид-ионов стабильность этих анионов уменьшается в ряду I¯>Br¯>Cl¯>F¯. Однако этот порядок соблюдается в апротонной среде. Напротив, в протонных растворителях или в присутствии кислотных катализаторов легче всего отщепляются анионы слабых кислот, поэтому порядок отщепления будет обратный (HF самая слабая кислота из всех галогеноводоводных кислот). Для этих реакций не требуются сильные нуклеофилы, но необходимо, чтобы субстраты имели хорошие уходящие группы, поэтому большинство мономолекулярных реакций протекает в кислой среде.
Для SN2-реакций природа уходящей группы заметного влияния на скорость не оказывает, так как лимитирующая стадия здесь — образование переходного состояния, а отщепление замещаемой группы происходит, как правило, быстро. Но такие группы, как OH, OR, NH2 удаляются с трудом, так как связь углерод–кислород или углерод–азот достаточно прочная.
Таким образом, влияние различных факторов на направление и скорость реакций нуклеофильного замещения можно свести к следующим основным положениям.
Факторы, способствующие протеканию SN1-реакции:
1) образование стабильного карбокатиона,
2) применение высокополярного протонного растворителя и кислотных катализаторов,
3) стабильность уходящей группы.
Нуклеофильность атакующей частицы существенного значения не имеет.
Факторы, способствующие протеканию SN2-реакции:
1) доступность электрофильного центра субстрата,
2) применение апротонного растворителя,
3) высокая нуклеофильность реагента.
Природа уходящей группы существенного значения не имеет.