

Astruc D. - Modern arene chemistry (2002)(en)
.pdf3.2 Reactions with Aryl Halides and Triflates: Synthesis of Biaryls 63
ð16Þ
dibromodihydroxytropolone derivatives and a variety of boronic acids [40]. An example is outlined in Eq. (17). These compounds were found to be potent inhibitors of inositol monophosphatase with IC50 values in the low-micromolar range.
ð17Þ
Under standard Suzuki reaction conditions, the coupling of tricarbonyl[h6-(diphenylphos- phino)benzene]chromium(0) with typical arylboronic acids has been achieved, giving biaryl complexes in high yield (Eq. (18)). This type of reaction sequence serves to generate product complexes that are not available by other means [41].
ð18Þ
Similar coupling reactions of other chromium(0) complexes with arylboronic acids have also been reported [42, 43].
Uemura and Kamikawa have presented a review on the stereoselective synthesis of axially chiral biaryls utilizing planar chiral (arene)chromium complexes [44].
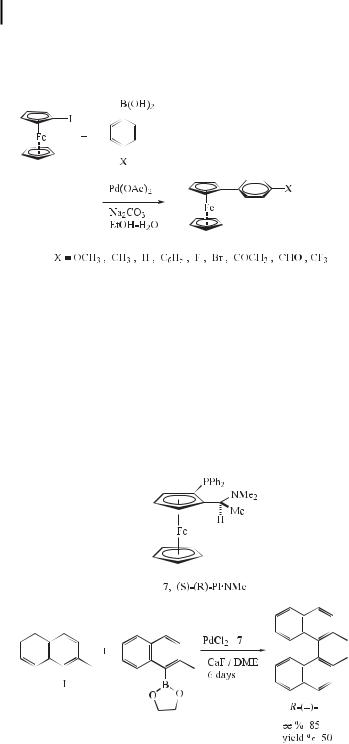
A modification of the Suzuki coupling reaction proved to be a clean and useful method for the preparation of monosubstituted arylferrocenes. Iodoferrocene was reacted with a series of substituted arylboronic acids in the presence of sodium carbonate and palladium acetate in aqueous ethanol at room temperature to produce a range of substituted monoarylferrocenes (Eq. (19)). A systematic investigation undertaken to determine the optimal reaction conditions indicated that scrupulous deoxygenation of the solvent is critical. The use of
643 The Suzuki Reaction with Arylboron Compounds in Arene Chemistry
stronger bases such as barium hydroxide and potassium carbonate is favorable and gives rise to better yields of monoarylferrocenes. The reactions also proceed e ciently in aqueous DMF, broadening the scope of the process and allowing e cient reactions with boronic acids that show low solubility in organic solvents [45].
ð19Þ
Ferrocenyl-substituted aldehydes have also been prepared by a Suzuki reaction [46]. Restricted rotation about the aryl–aryl bond in biaryls can lead to the phenomenon of
atropisomerism. Chiral binaphthalenes constitute an important class of such atropisomeric compounds, not least because they are among the most useful chiral ligands and auxiliaries employed in asymmetric synthesis. A small number of asymmetric biaryl syntheses have been reported. However, variable results have been reported depending on the nature of the catalyst used (e.g. Ni or Pd), and in all cases a Grignard reagent has been used as the organometallic component. Recently, Cammidge and Crepy [47] have reported examples of the use of an asymmetric Suzuki coupling protocol for the construction of binaphthalenes. Here, the ligand (7) with a tertiary amine moiety was found to lead to the highest selectivity, suggesting that the high ee obtained may stem from pre-complexation between the nitrogen in 7 and the boron prior to transmetalation (Eq. (20)).
ð20Þ

3.2 Reactions with Aryl Halides and Triflates: Synthesis of Biaryls 65
3.2.2
Aromatic–Heteroaromatic and Heteroaromatic–Heteroaromatic Couplings
Many Suzuki reactions have been applied in aromatic–heteroaromatic and heteroaromatic– heteroaromatic couplings by virtue of their synthetic utility in the pharmaceutical industry. This section deals with such reactions.
A one-step approach to heteroaryl benzoic acids from readily accessible heteroaryl halides and benzeneboronic acid esters has been reported. The coupling was carried out in the presence of Pd(PPh3)4 and sodium carbonate in aqueous acetonitrile (Eq. (21)) [48].
ð21Þ
For the synthesis of a series of 2,5-disubstituted pyrroles as selective dopamine D3 receptor antagonists, the preparation of 2-arylpyrroles as key intermediates was developed. As a first approach to 2-arylpyrroles, the reaction of Grignard reagents in a modification of a known reaction was tested. This method was, however, incompatible with the presence of acidic protons and basic nitrogens, and each desired substitution pattern required repetition of a lengthy reaction sequence. Finally, Johnson et al. [49] devised an alternative strategy based on palladium-catalyzed Suzuki aryl cross-coupling methodology, which showed promise as a short, direct, and more flexible route (Eq. (22)).
ð22Þ
The deep-red colored prodigiosin alkaloids are endowed with potent antibacterial, cytotoxic, and antimalaria properties. Additionally, in a series of recent reports it has been claimed that this family also displays a significant immunosuppressive activity. This finding is particularly noteworthy since the mechanism of action seems to be distinctly di erent from that of
663 The Suzuki Reaction with Arylboron Compounds in Arene Chemistry
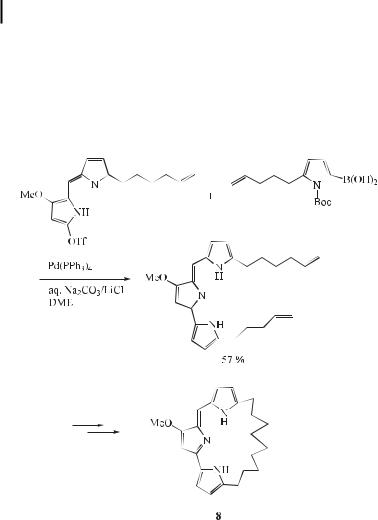
cyclosporin or FK-506. Consequently, these alkaloids are viewed as lead structures in the search for new drugs to prevent allograft rejection. Recently, the first total synthesis of the cyclic prodigiosin derivative 8 has been demonstrated (Eq. (23)) [50]. The key steps of this approach comprise a palladium-catalyzed Suzuki cross-coupling reaction of the rather unstable pyrroleboronic acid derivative with the electron-rich pyrrolyl triflate, followed by a ring-closing metathesis reaction.
ð23Þ
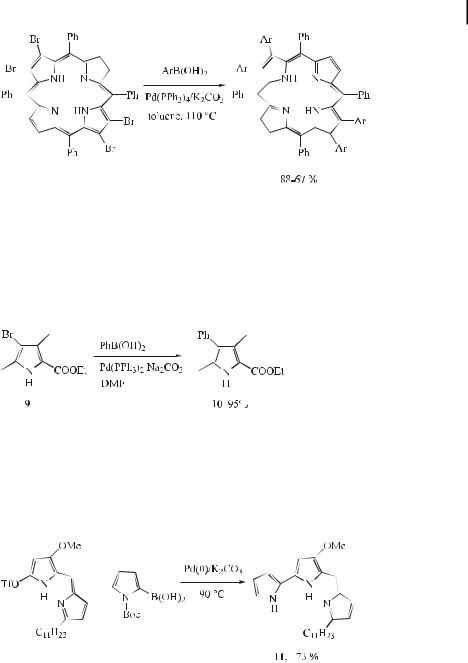
Porphyrin synthesis arouses continuing interest in biological, material, and inorganic chemistry. Substituents at the b-position of porphyrins exert much larger steric and electronic e ects on the porphyrin ring than substituents at the meso-aryl positions. However, the synthesis of b-substituted porphyrins often requires the relatively inaccessible 3-substituted or 3,4-disubstituted pyrroles for either protic or Lewis acid-catalyzed cotetramerization with aldehydes. Furthermore, in the preparation of unsymmetrical porphyrins, regioisomeric mixtures requiring di cult and tedious chromatographic purification are often obtained. Since b-brominated porphyrins are easily obtained by the controlled bromination of porphyrins or metalloporphyrins, the transformation of the bromo substituents into other functional groups would provide facile access to b-substituted porphyrins. Chan and co-workers have reported on the synthesis of b-aryl-substituted tetraphenylporphyrins by Suzuki cross-coupling of the corresponding b-bromoporphyrins (Eq. (24)) [51].
3.2 Reactions with Aryl Halides and Triflates: Synthesis of Biaryls 67
ð24Þ
Similarly, a facile synthesis of b-derivatized porphyrins has been reported by Nocera et al. [52].
In connection with such syntheses, Chang and Bag [53] also reported a synthetic route to tetramethyltetraphenylporphyrin from a pyrrole derivative. The required pyrroles were synthesized from bromopyrrole (9) and phenylboronic acid by means of a Pd(0)-catalyzed crosscoupling. The reaction proceeded well in DMF to give an essentially quantitative yield of the product 10 (Eq. (25)).
ð25Þ
Recently, undecylprodigiosine (11) was reported to inhibit T-cell proliferation at doses that are not cytotoxic, which is particularly attractive with regard to its potential clinical application. The hitherto published total syntheses of prodigiosines involve several steps and are not suitable for scale-up in the case of possible lead structure development. D’Alessio and Rossi [54] have devised a new synthetic pathway allowing the reproducible and consistent production of undecylprodigiosine (Eq. (26)).
ð26Þ
Non-steroidal antiinflammatory drugs (NSAIDs) are the main therapeutic agents for the treatment of the symptoms of arthritis. The drugs seem to inhibit the enzyme cyclooxygenase and consequently the conversion of arachidoic acid into prostaglandins. The main drawbacks of NSAIDs are severe side e ects, including gastrointestinal ulceration and
683 The Suzuki Reaction with Arylboron Compounds in Arene Chemistry
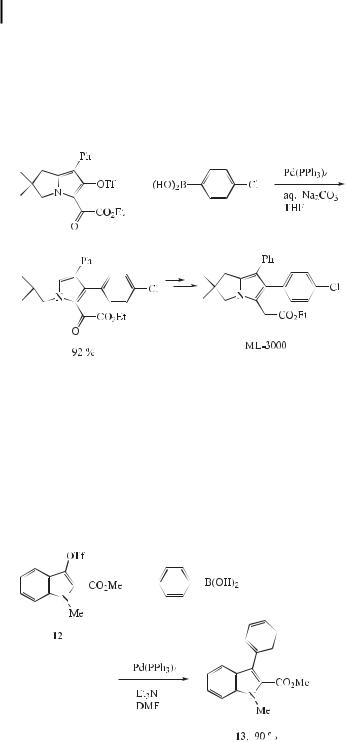
bronchospasm. Recently, 2,3-dihydro-1H-pyrrolizine derivatives such as ML-3000 were proven to selectively inhibit the enzymes cyclooxygenase and 5-lipooxygenase. Thus, ML3000 is one of the most potent and well-balanced dual inhibitors of both enzymes. However, the previous synthesis of ML-3000 proceeds with poor overall yields (<5 %). Most recently, Cossy and Belotti have reported that ML-3000 may be obtained from 1-chloro-3-phenyl-2- propyne in eight steps in an overall yield of 19 %. The key step is a Suzuki cross-coupling reaction between a heteroaryl triflate and (4-chlorophenyl)boronic acid (Eq. (27)) [55].
ð27Þ
In recent years, palladium(0)-catalyzed Suzuki coupling reactions have been reported in indole chemistry, whereby the indole moiety is usually introduced using an indolylboronic acid [56], but little work has been carried out with 2- or 3-haloindoles. Recently, MalapelAndrieu and Merour [57] reported such a Suzuki coupling using substituted 3-indolyltriflate and arylboronic acids, which a orded the corresponding 3-substituted indoles. Since 3- phenylindoles have interesting pharmacological structures, resembling endothelin antagonists, these authors have treated the triflate (12) with phenylboronic acid to produce the coupling product (13) in 90 % yield (Eq. (28)). Although it is well known that inorganic bases are recommended for the Suzuki reaction, organic bases such as triethylamine gave satisfactory results in this reaction.
ð28Þ
3.2 Reactions with Aryl Halides and Triflates: Synthesis of Biaryls 69
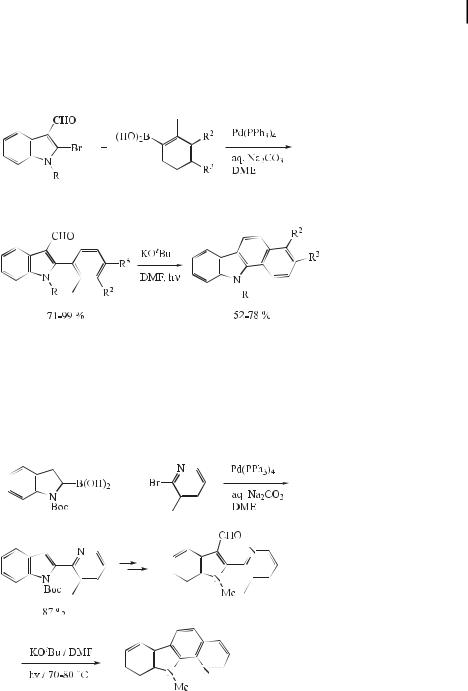
2-Bromoindole-3-carbaldehyde, or its N-methyl analogue, was found to undergo Suzuki coupling with boronic acids in the presence of palladium(0); subsequent treatment of the coupled products with potassium tert-butoxide gave the desired carbazoles in good yields (Eq. (29)) [58].
ð29Þ
A versatile and convenient method for the synthesis of substituted benzo[a]carbazoles and pyrido[2,3-a]carbazoles has recently been developed [59]. Treatment of 2-(o-tolyl)- or 2-(3- methyl-2-pyridyl)-substituted indole-3-carbaldehydes (obtained by Suzuki reaction) with potassium tert-butoxide in DMF at 70–80 C under irradiation by a 400 W high-pressure mercury lamp a orded benzo[a]carbazoles and pyrido[2,3-a]carbazoles, respectively, in good yields (Eq. (30)).
ð30Þ
In the course of an investigation on axial chirality in metal chelates [60], 4- and 5-aryl- substituted quinolin-8-ols were required. If, in the case of aryl groups bearing bulky sub-
703 The Suzuki Reaction with Arylboron Compounds in Arene Chemistry
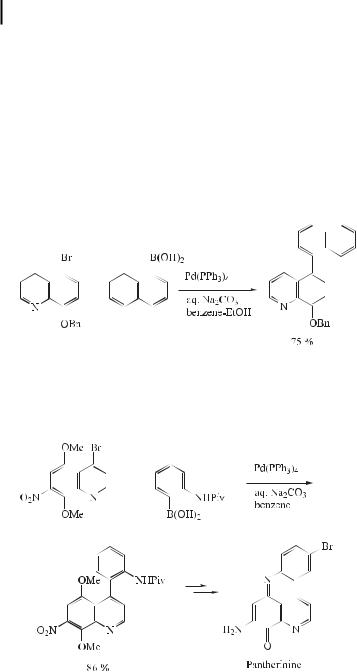
stituents on the quinoline ring, there is a su cient energy barrier to rotation about the pivotal bond, as in 5-arylquinolin-8-ols, this series of ligands is very fruitful for the development of a new entry to axial chirality based on quinolin-8-ol chelates. Furthermore, considering the applicability of these ligands in analytical science, electronic devices, and pharmaceuticals, they are evidently promising and important in various fields of science. Classically, 5-arylquinolin-8-ols have been prepared by a Skraup synthesis from 2-amino-4- arylphenols and acrolein or its equivalent. If it is possible to directly introduce the required aryl groups at the desired position of quinolin-8-ol, the synthetic route is much more advantageous. Recently, such a direct introduction of aryl groups was reported by Nakano [61], employing a Suzuki coupling reaction (Eq. (31)).
ð31Þ
During the last decade a series of structurally fascinating and biologically active fused polycyclic aromatic alkaloids has been isolated from marine sources. One such alkaloid, pantherinine, has recently been synthesized using the Suzuki reaction (Eq. (32)) [62].
ð32Þ
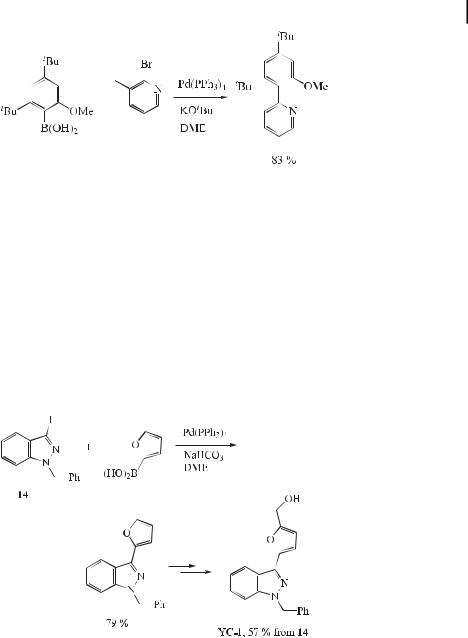
A highly sterically hindered pyridylphenol derivative has been synthesized by means of a Suzuki cross-coupling (Eq. (33)) [63].
3.2 Reactions with Aryl Halides and Triflates: Synthesis of Biaryls 71
ð33Þ
The indazole nucleus is a seldom used but e ective pharmacophore in medicinal chemistry, as illustrated by its application in pharmaceutical agents in fields as diverse as CNS disorders, antiinflammatories, and HIV protease inhibition. However, compared to indoles and benzimidazoles, indazole chemistry remains poorly studied due to the limited synthetic approaches to these compounds. Most syntheses of indazoles reported in literature start from benzene derivatives, and the pyrazole moiety is generated by ring-closure starting from isatins, o-substituted anilines, or phenyl hydrazines. These traditional synthetic routes do not readily facilitate substitution of at the 3-position with aryl or heteroaryl nuclei. Although YC- 1, a pharmacological agent with potential use in the treatment of cardiovascular diseases or erectile dysfunction, was obtained synthetically, its overall yield was only 4 % [64]. Recently, Rault et al. reported a new synthetic method for 3-arylindazoles based on a Suzuki-type coupling reaction of 3-iodoindazoles with aryland heteroarylboronic acids, which o ered a general and flexible route to 3-arylindazoles, including YC-1, in good yields (Eq. (34)) [65].
ð34Þ
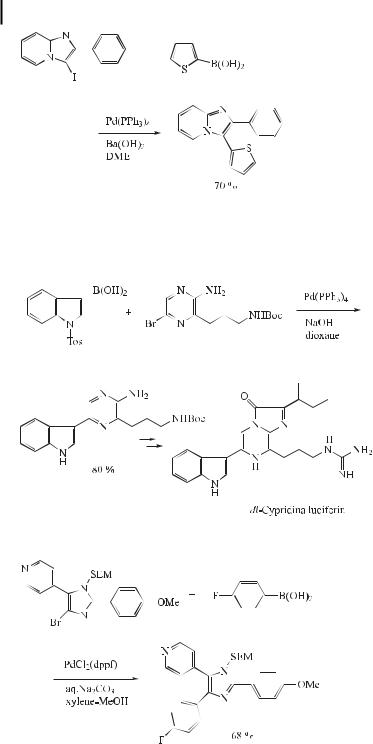
The influence of the base and solvent on Suzuki cross-coupling reactions of various 2- substituted 3-iodoimidazo[1,2-a]pyridines has been reported. The reactivity is mainly influenced by the nature of the substituent. Optimized yields and shortened reaction times were achieved using strong bases in DME (Eq. (35)) [66].
72 3 The Suzuki Reaction with Arylboron Compounds in Arene Chemistry
ð35Þ
Cypridina luciferin is an imidazopyrazinone involved in the bioluminescence of the crustacean Cypridina (Vargula) hilgendorfii. The first total synthesis of this natural product was achieved by Kishi in 1966, which was followed by that of White and Karpetsky in 1971. Most recently, Nakamura et al. developed a convenient, direct procedure for this synthesis, employing Suzuki coupling (Eq. (36)) [67].
ð36Þ
Biologically interesting 4,5-disubstituted and 2,4,5-trisubstituted imidazoles have been prepared by the Suzuki reaction (Eq. (37)) [68].
ð37Þ