

Astruc D. - Modern arene chemistry (2002)(en)
.pdfThe History of Benzene 3
August Kekule´ (1829–1892) is well known as one of the pioneers of modern structural theory in organic chemistry. He became interested in chemistry after attending classes of Justus von Liebig, then went to study in Paris with Charles Gerhardt, and became acquainted with JeanBaptiste Dumas. He enrolled in the University of Heidelberg as a Privatdozent in 1856, then in the
University of Ghent (Belgium) in 1858, and finally in the University of Bonn in 1867. His major contributions to chemistry were reiterating the tetravalency of carbon (first stated by A. S. Cooper), proposing its ability to form chains, and, of course, his drawing of the benzene structure that stimulated much synthetic work in aromatic chemistry.
Equilibrium proposed by Kekule´ (1872) to explain the unicity of ortho-disubstituted benzene
objection by suggesting that the two isomers of the disubstituted benzene are in rapid equilibrium, i.e. in what we now call a tautomeric equilibrium.
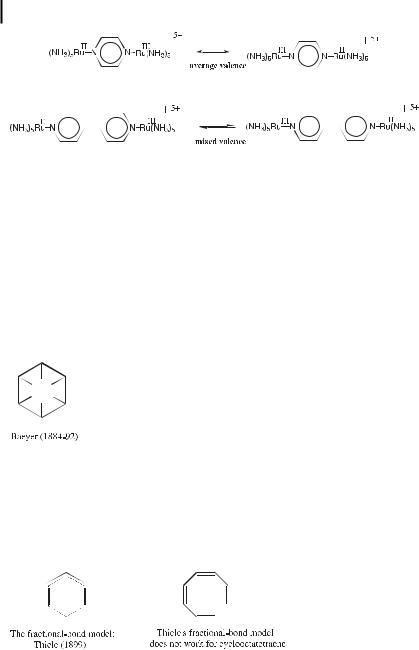
Although first-year students can now recognize Kekule´’s confusion, the answer was astute at that time, since the distinction between average valence and mixed valence would only be made a century later in 1969 by Henry Taube and Carol Creutz using Ru(II)-Ru(III) complexes [14]. In the 1930’s, Mills and Nixon evaluated this possibility with small rings being attached to benzene (vide infra).
4Arene Chemistry : From Historical Notes to the State of the Art
Another serious problem with Kekule´’s formula was the fact that cold KMnO4 and acids leave benzene unreacted whereas alkenes react with these reagents to give addition products. Indeed, Ladenburg showed experimentally in 1874 that the six carbon atoms of benzene are equivalent. After the main chemical properties of benzene had been established, the possible structure types became limited to the hexagon, the triangular prism, and the octahedron. Based on his experimental studies of benzene derivatives, Baeyer concluded that Claus’, Dewar’s, and Ladenburg’s formulae were untenable, and that benzene contained six carbon atoms in a ring, but did not accept Kekule´’s formula. He adopted a suggestion of Armstrong (1887) and proposed another hexagonal formula known as the Armstrong–Bayer centric formula, which had already been suggested by L. Meyer in 1865.
It is noteworthy that among the formulae proposed for benzene in the 19th century, only the first one, that of Loschmidt, is not far from being correct. The next acceptable formula only appeared with Thiele’s suggestion of fractional carbon–carbon bonds (partial valences) in 1899–1900. This formalism did not explain why cyclooctatetraene is not aromatic, however, as shown experimentally (for historical accounts, see refs. [15, 16]).
The name ‘‘benzene’’ was disputed in the 19th century. V. Meyer proposed ‘‘benzene’’ from ‘‘benzoin’’ because the means of preparing pure benzene involved decarboxylation of benzoic acid using sodium hydroxide at high temperature (otherwise, thiophene was an impurity that proved di cult to remove). Benzoic acid was obtained from gum benzoin as a white powder. On the other hand, Auguste Laurent, who inter alia taught crystallography to Louis

The History of Aromaticity 5
Pasteur in Paris and was one of the pioneers of modern atomic theory, proposed the name ‘‘phene’’. The word phene (from the Greek ‘‘phainen’’, to shine) was proposed because benzene burns with a bright flame. Although it was not adopted for benzene, this word is now used in a number of names of arenes, such as phenol, phenanthrene, etc.
The X-ray crystal structure of benzene, proving the equivalence of the six CaC bonds, appeared in 1929 and 1932, and Pauling reported its electron-di raction data in 1931. Note that several of the structures proposed in the 19th century, such as Dewar benzene (non-planar) and Ladenburg’s prismane, which are valence isomers of benzene, have now actually been prepared from benzene derivatives photochemically. They are kinetically stabilized, since they do not spontaneously revert to benzene or its derivatives [17–20].
The History of Aromaticity [21–32]
At the beginning of the 19th century, the compounds that were said to be ‘‘aromatic’’ were those having an aromatic smell. When the arenes were synthesized or isolated later in that century, the tendency was to distinguish two groups: the aromatic and non-aromatic (aliphatic, etc.) derivatives. The analysis of aromatic compounds showed unsaturation, although these compounds were di erent from alkenes and alkynes. In 1910, Pascal showed that aromatic compounds had exalted diamagnetic susceptibilities, and, in 1925, Armit and Robinson [33] suggested the aromatic sextet. The development of wave mechanics by Schro¨- dinger [34, 35] in 1926 led to molecular orbital theory, application of which led Hu¨ckel in 1931 to the fundamental idea of the p-electron molecular orbital and the well-known (4n þ 2) rule for aromatics and of anti-aromaticity for planar conjugated rings containing 4n p electrons [6, 7]. At about the same time, the theory of resonance, proposed by Slater [9], was based on the combinatorial representation of all the p electrons around the s skeleton. Thus, for benzene, among all the possible combinations, five were found to provide the best contribution to the real structure, i.e. two Kekule´-type and three Dewar-type structures, which
Canonical valence-bond structures of benzene according to the theory of resonance by Slater (1929)
were termed ‘‘canonical’’ valence-bond structures. This valence-bond approach with its structural representation was exploited by Pauling and became commonly used [11, 36].
Pauling proposed the theory of ring currents in 1935, i.e. free electron circulation around the benzene ring. In the following year, 1936, London stated that the p-electron circulation is responsible for a diamagnetic contribution to magnetic susceptibility. These ring-current effects on NMR chemical shifts were disclosed by Pople in 1956 [37]. By the end of the 1960s, the development of molecular orbital theory had extended to non-benzenoid compounds [15, 38]. Modern theories taking into account the exaltation and anisotropy of magnetic susceptibility by Dauben and Flygare appeared at the end of the 1960s, and quantum chemical calculations were reported by Kutzelnig in 1980.

6Arene Chemistry : From Historical Notes to the State of the Art
The criteria for aromaticity have been numerous and have changed with time. Chemical reactivity was the only criterion of aromaticity at the end of the 19th century, since a good number of electrophilic substitution reactions of arenes were already known. Thus, aromatic systems reacted with bromine to give the substitution product with retention of the aromatic character, whereas non-aromatic unsaturated compounds readily added bromine to form a dibromide. This distinction was used as a guide to define aromatic and non-aromatic compounds. This easy rule-of-thumb criterion for neutral compounds has obviously been very useful owing to its simplicity, and has survived to the present day. It is by no means general, however. For instance, anthracene and phenanthrene add bromine, and the former is used as a diene in Diels–Alder reactions. Ferrocene, a super aromatic, does not react with bromine to give the substitution product, but rather the bromide salt of the oxidized ferrocenium cation [39]. Fullerenes cannot give substitution products upon reaction with bromine, but addition products are readily obtained. Thus, fullerenes show a non-aromatic, olefin-like behavior, yet they are somewhat aromatic, although much less so than planar arenes [40].
A classical criterion that was frequently mentioned at the beginning of the 20th century was the thermodynamic one. Since DH for the hydrogenation of cyclohexene is 120 kJ mol 1, the hypothetical cyclohexatriene should have a DH of hydrogenation of 360 kJ mol 1. Experimentally, DH for the hydrogenation of benzene amounts to just 210 kJ mol 1, a di erence of 150 kJ mol 1, suggesting that benzene is more stable than the hypothetical cyclohexatriene by 150 kJ mol 1. This stabilization energy may be thought of as an approximation of the resonance energy. There are uncertainties associated with the approximation made in the comparisons, however, and theoretical calculation led to an estimate of 40–120 kJ mol 1 for the resonance energy of benzene. Thus, this criterion is not satisfactory, especially if one tries to extend it to other arenes and heteroarenes.
In 1959, Albert suggested that the criterion of aromaticity should be based on bond lengths [38]. Thus, a ring was classed as aromatic if its CaC bond lengths were the same as those in benzene. A refinement was that the molecule was deemed aromatic if its CaC bond
|
˚ |
lengths were between 1.36 and 1.43 A, while it was deemed a polyene if it had alternat- |
|
˚ |
˚ |
ing bond lengths of 1.34 to 1.356 A for the double bonds and 1.44 to 1.475 A for single
bonds. Another refinement was based on the mean-square deviations of the CaC bond lengths as a quantitative criterion for measuring of aromaticity. This definition was not very convenient because, in most cases, accurate bond lengths were unknown, and it did not apply to heteroatom-containing systems. Also, some well-known aromatics have some long bonds (for instance, the transannular bond of azulene). Thus, this criterion is not generally rigorously applicable. A further example for which it fails is borazine, which, despite equal bond lengths, is not aromatic. The bond lengths in the cyclopentadienyl cation (0.1425 nm, anti-aromatic) and in the cyclopentadienyl anion (0.1414 nm, aromatic) are also almost identical [41]. The magnetic ring current e ects constitute the modern criterion of aromaticity that is now considered as the most reliable one. Their experimental e ects are (i) the well-known anomalous chemical shifts in 1H NMR, (ii) large magnetic anisotropies, and (iii) diamagnetic susceptibility exaltation. The mobile electrons are not only p electrons, but can also be s or mixed, as has long been exemplified by the transition states of electrocyclic reactions. While the high field 1H NMR chemical shifts found for aromatic protons on the exterior of an aromatic ring o er a straightforward, easy, and popular criterion, there are complications. For instance, with [16] annulene, the inner and outer protons resonate in the

The History of Aromaticity 7
opposite sense from what would be expected based on simple arguments because [4n] annulenes have low-lying excited states leading to a large paramagnetic e ect that reverses ring currents [41].
Prototypes of Sondheimer’s annulenes (1956–7)
Thus, attempts to make the e ect of ring current on 1H NMR chemical shifts a quantitative criterion have met with serious problems, suggesting that other e ects may interfere. The large diamagnetic susceptibility exaltation D, however, is uniquely associated with aromaticity, whereas anti-aromatic compounds exhibit paramagnetic exaltation. The diamagnetic susceptibility exaltation D is defined as the di erence between the bulk magnetic susceptibility wM of a compound and the susceptibility wM0 estimated from an increment system for the structural components such as isomers without cyclic delocalization (D ¼ wM wM0). The experimental results have recently been satisfyingly compared with computational data by von Rague´ Schleyer [41]. Thus, the definition of aromatic compounds as those exhibiting a significantly exalted diamagnetic susceptibility now appears to be an absolute criterion [37].
Anti-aromaticity was predicted by the Hu¨ckel approach for conjugated cyclic planar structures with 4n p electrons due to the presence of two electrons in antibonding orbitals, such as in the cyclopropenyl anion, cyclobutadiene, and the cyclopentadienyl cation (n ¼ 1), and in the cycloheptatrienyl anion and cyclooctatetraene (n ¼ 2). It has been argued that a simple definition of an anti-aromatic molecule is one for which the 1H NMR shifts reveal a paramagnetic ring current, but the subject is controversial. The power of the Hu¨ckel theory indeed resides not only in the aromatic stabilization of cyclic 4n þ 2 electron systems, but also in the destabilization of those with 4n electrons [22, 27, 42].
The term homoaromaticity has been coined by Winstein [43, 44]. The rupture of a cyclic conjugation due to the insertion of a saturated fragment such as CH2 partly preserves the aromatic stabilization of the original aromatic molecule or ion. Winstein suggested that homoaromaticity, a type of aromaticity, is found for cations that have neither the s-electron
8Arene Chemistry : From Historical Notes to the State of the Art
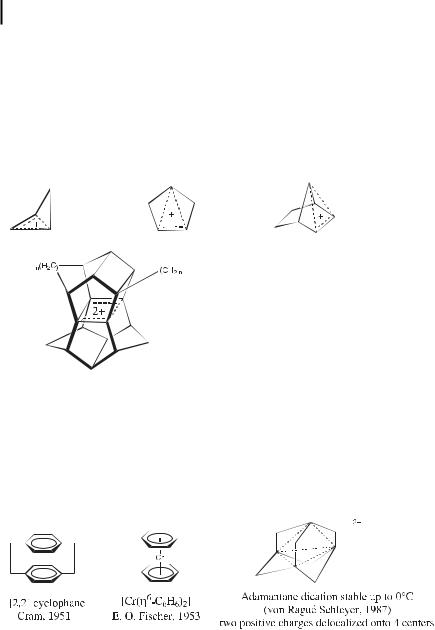
framework of a classical aromatic system nor a parallel p-orbital arrangement, but that have an arrangement resembling the cyclic one with (4n þ 2) electrons. For instance, the cyclobutenyl cation is homoaromatic, because it resembles the cyclopropenyl cation. Remarkably, most examples of homoaromaticity are those of cations in which the pp atomic orbitals involved are not parallel, the prototype being the norbornenyl cation, which has a ‘‘bishomoaromatic’’ arrangement. Indeed, a key experiment concerning homoaromaticity was the solvolysis of the anti-7-norbornenyl tosylate, which was found to be 1011 times faster than that of the saturated analogue [24]. Prinzbach’s pagodane dications [45] are inter alia [44] remarkable examples.
Prinzbach’s 4-center 2-electron bis-homoaromatic pagodane dication
Three-dimensional aromaticity has been demonstrated, for instance in the case of Cram’s cyclophane in 1951 [46, 47] and, more recently, for von Rague´ Schleyer’s adamantane dication [48], half-sandwich carbocations, and nido-carboranes. Ferrocene and various other organometallic compounds also are three-dimensional aromatics. ‘‘Spherical aromaticity’’ is exhibited by closo-carboranes, borane anions, and, to a lesser extent, C60, although these compounds are isotropic.
Tridimensional aromaticity
In [m] circulenes, a family of polyaromatic hydrocarbons so named in 1975 by Wynsberg, in which m refers to the number of aromatic rings arranged in a circle, the total number of p electrons does not indicate aromaticity or anti-aromaticity according to the Hu¨ckel rule. This rule is strictly only applicable to monocyclic systems. It is adequate, however, to consider the inner and the outer p electrons separately, whose numbers obey the 4n þ 2 Hu¨ckel criterion for aromaticity, since both these circuits are monocyclic [49]. Coronene, a flat graphite frag-

Some Key Trends Towards Modern Arene Chemistry 9
ment of D6h symmetry synthesized in 1932 is the prototype of circulene with m ¼ 6 [50]. It would formally be anti-aromatic according to the Hu¨ckel rule, since it contains 24 p electrons. The inner and outer sub-circuits contain 6 and 18 p electrons, respectively, however, and these numbers conform to the aromatic 4n þ 2 series. The same situation arises for corannulene (m ¼ 5), a bowl-shaped hydrocarbon with D5h symmetry corresponding to onethird of C60, that was first synthesized by Schott and Meyer in 1966 [51]. It has 20 p electrons, which would also correspond to anti-aromaticity. The inner ring can be viewed as an aromatic cyclopentadienyl anion with 6 p electrons, however, leaving 14 electrons for the cationic outer sub-circuit, which also correspond to 4n þ 2 electrons with n ¼ 3. Pleiadannulene, the [7] circulene synthesized by Yamamoto in 1983 [52], has a saddle-shaped structure with static C2 symmetry and dynamic D7h symmetry. It would also formally be antiaromatic with 28 p electrons (4n; n ¼ 7). However, the inner cycloheptatrienyl cation (6 p electrons) and outer anion sub-circuit (22 p electrons) can be viewed as aromatic fragments since they obey the 4n þ 2 rule of Hu¨ckel. The most popular higher circulene is kekulene, a planar hydrocarbon with D6h symmetry first synthesized in 1978 by Diederich and Staab, which is also called circumcoronene because its inner ring is equivalent to the outer ring of coronene [53].
[m]-Circulenes viewed with a central aromatic (be) core and an aromatic (4n þ 2e) annulene outer circuit
10 Arene Chemistry : From Historical Notes to the State of the Art
Higher PAHs have also been the subject of theoretical studies, including graph theories [54, 55]. The idea of Loschmidt on the representation of benzene using annotated circles, ascribing elemental character to the ring of six carbons in benzene has been revived by Siegel, who used the point replacement for benzene called the Loschmidt replacement. A very large variety of modern arene structures were shown using this representation [56].
Some Key Trends Towards Modern Arene Chemistry
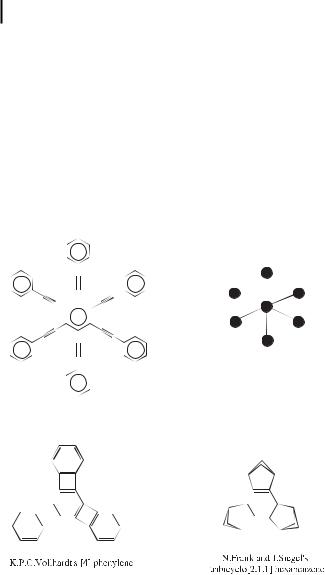
Attempts to localize the double bonds of the benzene ring in a Kekule´-like structure have been made with some success by the groups of Vollhardt [57] and of Frank and Siegel [58]. This was reminiscent of the Mills–Nixon e ect predicted in the 1930’s, although the partial localization has nothing to do with the tautomeric equilibrium in benzene argued by Kekule´.
‘‘Loschmidt-replacement’’ representation of hexaalkynylbenzene
Threefold-symmetric arene structures synthesized with the aim of localizing the double bonds in the benzene ring
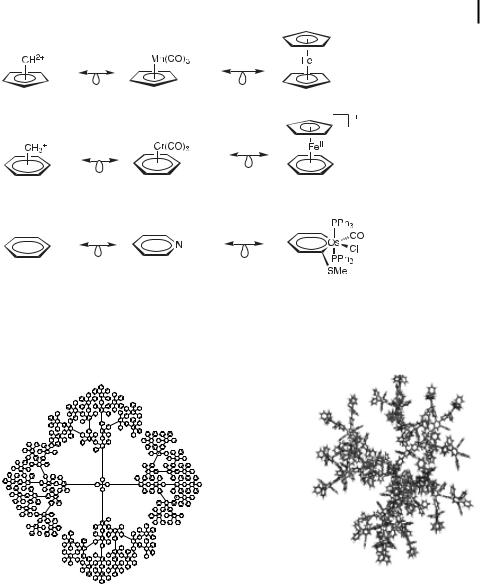
The aromatic families have benefited from Ho mann’s concepts of isolobal analogy that links organic fragments to inorganic ones [59].
Besides these classical aromatics and polyaromatic hydrocarbons, other very important classes or arene molecules are: porphyrins [60, 61], phthalocyanins [61, 62], porphycens [63], calixarenes [64], resorcarenes [64], cyclophanes [47], dendrimers [65], elementa-arenes [66], organometallic arene (hexahapto) [67], benzyne (dihapto), and aryland benzyl (monohapto) complexes [68], inorganic pyridine and polypyridine complexes [69], fullerenes [70, 71], and
Aromatic Chemistry : From the 19th Century Industry to the State of the Art 11
Isolobal analogy between organic and organometallic arene and cyclopentadienyl fragments and compounds (R. Ho mann, 1975)
carbon nanotubes [72]. Arene complexes that are not aromatic have the tetrahapto and dihapto coordination modes, and also include trihapto and pentahapto benzyl complexes [68].
Mu€llen’s fourth generation polyphenylene dendrimer [65]
Aromatic Chemistry : From the 19th Century Industry to the State of the Art
From a synthetic standpoint, a historical landmark, after the discovery of electrophilic substitution in the 1860’s, was the synthesis of aspirin, acetylsalicylic acid. The earliest known use of the drug can be traced back to the Greek physician Hippocrates in the 5th century BC. He used powder extracted from the bark of willow trees to treat pain and reduce fever. Salicin, the parent of the salicylate drug family that generates salicylic acid in vivo, was isolated
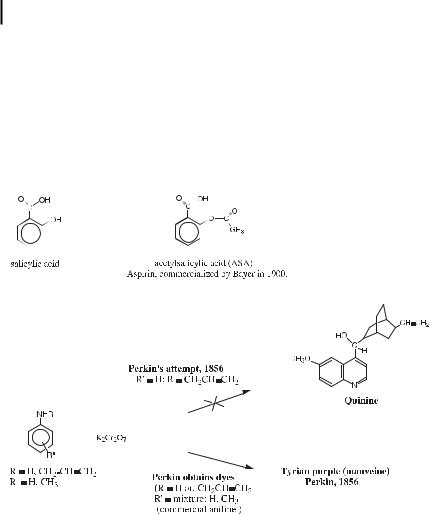
12Arene Chemistry : From Historical Notes to the State of the Art
in 1829 from willow bark. Sodium salicylate, the predecessor of aspirin was developed in 1875 as a pain reliever. This drug caused stomach irritation, however. In 1897, Felix Ho - man, a German chemist working for Bayer, who had given sodium salicylate to his father to treat arthritis and encountered the same problem, found a new synthesis of a less acidic formula, acetylsalicylic acid (ASA), which was patented in 1899 and commercialized by Bayer in 1900. The e ect of aspirin was not understood until 1970 when the British pharmacologist John Vane found that it inhibited the release of prostaglandin. Aspirin regulates blood vessel elasticity and changes the functions of blood platelets, and therefore has also become a popular drug to prevent heart attacks and circulatory troubles.
Perkin finds Tyrian purple (mauveine), and this sets the start of the industial chemistry with dyes
Industrial aromatic chemistry was initiated in the 1850s with the pioneering e orts of William H. Perkin in London, where he started to work as a student of August W. von Ho mann, himself a bright student of Justus von Liebig [73]. During attempts to synthesize quinine, the only substance known, at that time, to be e ective against malaria, he obtained dyes, and started to develop that field pertaining to aromatic chemistry.
With the influence of an artist friend interested in painting and dyes, he started to develop aniline purple (Tyrian purple) production, which he patented in 1856 and set up a factory for. The dyestu industry soon flourished. In 1859, Emanuel Verguin prepared the important dye fuschine, which was subsequently produced in Basel. In 1869, Perkin patented the synthesis of the natural dye alizarin, at the same time as Caro, Lieberman, and Graebe did so in Germany. Later, moreover, Perkin prepared alizarin from anthracene, which had been