2.8.16. Сухой остаток экстрактов
В плоскодонную чашку диаметром около 50 мм и высотой около 30 мм быстро вносят 2,00 г или 2,0 мл испытуемого экстракта. Выпаривают досуха на водяной бане и сушат при температуре (100 - 105) °С в течение 3 ч. Охлаждают в эксикаторе над фосфора (V) оксидом Р или силикагелем безводным Р и взвешивают. Результат рассчитывают как массовый процент или в граммах на литр.
# Допускается использовать в качестве осушающего агента кальция хлорид безводный Р.
2.8.17. Потеря в массе при высушивании экстракта
В плоскодонную чашку диаметром 50 мм и высотой 30 мм быстро взвешивают 0,50 г мелко измельчённого испытуемого экстракта. Сушат при температуре (100 - 105) °С в течение 3 ч. Охлаждают в эксикаторе над фосфора (V) оксидом Р или силикагелем безводным Р и взвешивают. Результат рассчитывают в массовых процентах.
# Допускается использовать в качестве осушающего агента кальция хлорид безводный Р.
# 2.8.18. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЭКСТРАКТИВНЫХ ВЕЩЕСТВ
Около 1 г измельчённого сырья (точная навеска), просеянного сквозь сито с отверстиями диаметром 1 мм, помещают в коническую колбу вместимостью 200-250 мл, прибавляют 50 мл растворителя, указанного в нормативной документации на лекарственное сырьё, колбу закрывают пробкой, взвешивают (с погрешностью ±0,01 г) и оставляют на 1 ч. Затем колбу соединяют с обратным холодильником, нагревают, поддерживая слабое кипение, в течение 2 ч. После охлаждения колбу с содержимым вновь закрывают той же пробкой, взвешивают и потерю в массе восполняют растворителем. Содержимое колбы тщательно взбалтывают и фильтруют через сухой бумажный фильтр в сухую колбу вместимостью 150-200 мл. 25 мл фильтрата пипеткой переносят в предварительно высушенную при температуре (100 - 105)°С до постоянной массы и точно взвешенную фарфоровую чашку диаметром 7-9 см и выпаривают на водяной бане досуха. Чашку с остатком сушат при температуре (100 - 105)°С до постоянной массы, после чего охлаждают в течение 30 мин в эксикаторе, на дне которого находится хлорид кальция безводный Р, и немедленно взвешивают.
Проводят два параллельных определения.
Содержимое экстрактивных веществ в процентах в пересчёте на абсолютно сухое сырьё вычисляют по формуле:
М 200 100
М
• (100 - W У
где:
м - масса сухого остатка в граммах;
м1 - масса сырья в граммах;
W - потеря в массе при высушивании сырья в процентах.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений.
Для спирторастворимых веществ рекомендуется использовать растворитель с концентрацией, обозначенной в методике испытаний на конкретный вид лекарственного растительного сырья; для водорастворимых веществ рекомендуется использовать в качестве растворителя воду. Возможно также использование других растворителей, указанных в методике испытаний.
# 2.8.19. ПРАВИЛА ПРИЁМКИ И МЕТОДЫ ОТБОРА ПРОБ
Отбор проб лекарственного растительного сырья «ангро»
Приёмку лекарственного растительного сырья производят партиями.
Партией считают любое количество цельного, обмолоченного, измельченного, прессованного лекарственного растительного сырья, однородного по способу подготовки и показателям качества, одного наименования и оформленного одним документом, удостоверяющим его качество. Документ, удостоверяющий качество партии должен содержать следующие данные:
номер и дату выдачи документа;
наименование и адрес отправителя;
наименование сырья;
номер партии;
массу партии;
год и месяц сбора или заготовки;
район заготовки (для сырья из дикорастущих растений);
результаты испытаний качества сырья;
наименование нормативной документации на сырьё;
подпись лица, ответственного за качество сырья, с указанием фамилии и должности.
Каждую единицу продукции подвергают внешнему осмотру для установления соответствия упаковки и маркировки требованиям нормативной документации. Обращают внимание на правильность упаковки, состояние тары (отсутствие подмочки, подтеков и других повреждений, отрицательно влияющих на качество и сохранность сырья).
Для проверки соответствия качества сырья требованиям нормативной документации отбирают выборку из неповрежденных единиц продукции, взятых из разных мест партии в количестве, указанном в табл. 2.8.19.-1. Проверку качества сырья в поврежденных единицах продукции производят отдельно от неповрежденных, вскрывая каждую единицу продукции.
|
№ п/п |
Количество единиц продукции сырья |
Объём выборки |
|
1 |
1 - 5 |
Все транспортные единицы |
|
2 |
6 - 50 |
5 транспортных единиц |
|
3 |
Свыше 50 |
10 % транспортных единиц от партии |
Примечание. Неполные 10 единиц продукции приравнивают к 10 единицам (например, при наличии в партии 51 единицы продукции объём выборки составляет 6 единиц).
Попавшие в выборку единицы продукции вскрывают и путем внешнего осмотра определяют: однородность сырья по способу подготовки (цельное, измельченное, прессованное и т. д.), цвету, запаху, засоренности; наличие недопустимых примесей -плесени, гнили, устойчивого постороннего запаха, не исчезающего при проветривании; засоренность ядовитыми растениями и посторонними примесями (камни, стекло, помет грызунов и птиц и т.д.). Одновременно невооруженным глазом и с помощью лупы (5— 10 X) определяют наличие амбарных вредителей
При установлении (внешний осмотр) неоднородности сырья, наличия плесени и гнили, засоренности посторонними растениями в количествах, явно превышающих допустимые примеси и т. д. вся партия должна быть рассортирована, после чего вторично предъявлена к сдаче.
При обнаружении в сырье затхлого, устойчивого постороннего запаха, не исчезающего при проветривании, ядовитых растений и посторонних примесей (помет грызунов и птиц, стекло и др.), зараженности амбарными вредителями II и III степеней, партия сырья не подлежит приемке.
Из каждой единицы продукции, отобранной для вскрытия, берут, избегая измельчения, 3 точечные пробы: сверху, снизу и из середины. Из мешков, тюков и кип точечные пробы отбирают на глубине не менее 10 см рукой сверху, затем, после распарывания по шву, из середины и снизу; точечные пробы семян и сухих плодов отбирают зерновым щупом. Из сырья, упакованного в ящик, первую точечную пробу отбирают из верхнего слоя, вторую - после удаления сырья примерно до половины ящика и третью - со дна ящика. Точечные пробы должны быть примерно одинаковыми по массе. Из всех точечных проб, осторожно перемешивая, составляют объединенную пробу.
В случае если масса объединённой пробы недостаточна для проведения всех испытаний, отбор точечных проб повторяют.
Из объединённой пробы методом квартования выделяют следующие пробы в приведённой ниже последовательности:
пробу для определения степени зараженности амбарными вредителями массой 500 г для мелких видов сырья и массой 1000 г для крупных видов сырья;
пробу для определения подлинности, измельченности и содержания примесей в соответствии с таблицей 2.8.19-2.
Для этого лекарственное растительное сырьё разравнивают на гладкой, чистой, ровной поверхности в виде квадрата по возможности тонким равномерным по толщине слоем и по диагонали делят на четыре треугольника. Два противоположных треугольника удаляют, а два оставшихся соединяют вместе и перемешивают. Эту процедуру повторяют до тех пор, пока не останется количество сырья в двух противоположных треугольниках, соответствующее массе заданных проб. Допустимые отклонения в массе каждой из проб не должны превышать ±10%.
Оставшуюся часть объединенной пробы измельчают ножницами или секатором до размеров частиц около 1 см, перемешивают и выделяют методом квартования аналитические пробы для определения:
влажности (сразу после отбора пробу упаковывают герметически) в соответствии с таблицей 2.8.19-2;
золы и действующих веществ в соответствии с таблицей 2.8.19-2;
микробиологической чистоты массой 50 - 200 г;
радионуклидов в соответствии с указаниями таблицы 2.8.19.-3.
пестицидов и токсических веществ массой 1 кг
|
Наименование сырья |
Масса пробы г, для определения | ||
|
Подлинности, измельчённости и содержания примесей |
Влажности |
Содержания золы и действующих веществ | |
|
почки березовые |
50 |
25 |
25 |
|
почки сосновые |
200 |
25 |
100 |
|
листья цельные, кроме нижеперечисленных: |
200 |
25 |
150 |
|
лист сенны |
100 |
15 |
50 |
|
лист толокнянки и брусники |
50 |
25 |
50 |
|
листья резаные, обмолоченные |
5 |
25 |
100 |
|
цветки, кроме нижеперечисленных: |
200 |
25 |
50 |
|
цветки полыни цитварной |
25 |
15 |
50 |
|
цветки ноготков, кукурузные столбики с рыльцами |
100 |
25 |
50 |
|
цветки бузины черной |
20 |
15 |
25 |
|
цветки ромашки аптечной |
50 |
25 |
100 |
|
цветки ромашки далматской |
300 |
25 |
50 |
|
травы цельные, побеги, кроме нижеперечисленных: |
300 |
50 |
200 |
|
трава душицы |
25 |
15 |
50 |
|
побеги анабазиса |
50 |
25 |
100 |
|
травы резаные, обмолоченные |
50 |
25 |
100 |
|
сочные плоды, кроме нижеперечисленных: |
100 |
50 |
50 |
|
плоды шиповника |
200 |
25 |
50 |
|
плоды стручкового перца |
300 |
25 |
150 |
|
сухие плоды и семена, кроме нижеперечисленных: |
200 |
25 |
50 |
|
семена дурмана индейского, термопсиса, льна |
50 |
25 |
100 |
|
плоды амми и семена джута |
10 |
25 |
100 |
|
клубни, корни и корневища цельные, кроме нижеперечисленных: |
300 |
50 |
200 |
|
корневище и корень марены, корневище лапчатки |
200 |
50 |
100 |
|
клубни салепа |
100 |
25 |
50 |
|
корневище и корень девясила |
600 |
50 |
100 |
|
корневище мужского папоротника и корень ревеня |
1000 |
100 |
300 |
|
корень мыльный туркестанский |
10000 |
200 |
- |
|
корень солодки очищенный |
5000 |
100 |
500 |
|
корень солодки неочищенный, корень барбариса |
2000 |
100 |
200 |
|
корни и корневища резаные, дробленые |
100 |
25 |
100 |
|
Корни и корневища в порошке |
50 |
15 |
25 |
|
кора цельная |
400 |
50 |
100 |
|
кора резаная |
100 |
25 |
50 |
|
прочее растительное сырье: |
|
|
|
|
ликоподий |
50 |
25 |
25 |
|
|
|
|
|
|
50 |
25 |
100 |
|
|
березовый гриб — чага |
2000 |
500 |
100 |
|
морская капуста — слоевища |
3000 |
500 |
1000 |
|
морская капуста шинкованная |
500 |
100 |
300 |
|
морская капуста — порошок |
100 |
50 |
200 |
|
сырье животного происхождения: |
|
|
|
 рожки
спорыньи
рожки
спорыньибадяга10025-Пробу для установления степени зараженности амбарными вредителями помещают в плотно закрывающуюся ёмкость. Пробы для определения радионуклидов, пестицидов, токсических веществ и микробиологической чистоты упаковывают каждую в полиэтиленовый или многослойный бумажный пакет. К пакету или емкости прикрепляют этикетку, такую же этикетку вкладывают внутрь мешка или емкости. На этикетке указывают следующие данные: наименование сырья; наименование поставщика; номер партии; массу партии; дату отбора пробы, фамилию и должность лица, отобравшего пробу.
Аналитические пробы должны быть взвешены с погрешностью ±:
0,01 - при массе пробы до 50 г;
0,1 - при массе пробы 50 - 500 г;
1,0 - при массе пробы 50 - 1000 г;
5,0 - при массе пробы более 1000 г.
При установлении в результате испытаний несоответствия качества сырья требованиям нормативной документации проводят его повторную проверку. Для повторного анализа от невскрытых единиц продукции отбирают выборку в соответствии с таблицей 2.8.19.-1. Результаты повторного анализа являются окончательными и распространяются на всю партию.
После отбора проб составляется акт отбора образцов, в котором указывается:
Наименование лекарственного растительного сырья;
Номер партии;
Масса партии ;
Дата отбора проб;
Фамилия лица, отбиравшего пробы
Примечание. Отбор проб корня женьшеня осуществляется в соответствии с частной фармакопейной статьёй.
Отбор проб фасованной продукции
Лекарственное растительное сырье и сборы расфасовываются в пачки, пакеты, фильтр-пакеты в цельном, резаном, дробленом, порошкованном, резано-прессованном виде, а также в форме брикетов для использования в качестве лекарственных средств.
Приемку фасованной продукции проводят сериями. Под серией понимают определённое количество однородного по всем показателям фасованного лекарственного растительного средства (цельное, измельченное, порошок), произведённое в течение одного технологического цикла, оформленное одним документом качества. Серия формируется из одной или нескольких партий лекарственного растительного сырья.
Единицы продукции в выборку необходимо отбирать из разных мест контролируемой серии.
где:
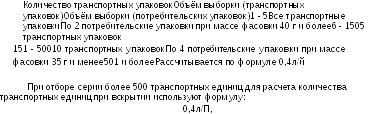 Объем
выборки зависит от объема серии и указан
в таблице 2.8.19.-4.
Объем
выборки зависит от объема серии и указан
в таблице 2.8.19.-4.
n
количество упаковочных единиц в одной серии.
Полученное в результате подсчёта по формуле дробное число округляют в сторону увеличения до целого числа, оно должно быть не менее 3 и не более 30. В случае недостаточного количества упаковочных единиц для проведения испытания, повторно отбирают упаковочные единицы, как указано выше.
Отобранные потребительские упаковки составляют объединённую пробу.
Из объединённой пробы выделяются пробы для определения:
допустимых отклонений на промышленное фасование - 10 невскрытых пачек или пакетов, 10 невскрытых контурных ячейковых упаковок, брикетов, 10 невскрытых пачек с фильтр-пакетами;
микробиологической чистоты - 5 невскрытых потребительских упаковок общей массой не менее 50 г;
радионуклидов в соответствии с таблицей 2.8.19.-5
аналитические пробы в соответствии с таблицей 2.8.19.-2
пестицидов и токсических веществ - 10 невскрытых потребительских упаковок общей массой не менее 500 г;
|
Количество потребительских упаковок, шт. |
Объём выборки, шт. |
|
До 100 |
2 (но не менее 70 г) |
|
От101 до 200 |
3 (но не менее 70 г) |
|
От 201 до 500 |
4 (но не менее 70 г) |
От 501 и более
5 (но не менее 70 г)
Отобранные упаковки объединённой пробы, после выделения проб для определения микробиологической чистоты, пестицидов, токсических веществ и отклонения в массе, вскрывают, содержимое высыпают на гладкую, чистую, ровную поверхность, тщательно перемешивают и методом квартования выделяют пробы, соответствующие по массе одной из заданных проб (таблица 2.8.19.-2).
Определение допустимых отклонений на промышленное фасование определяют следующим образом.
Анализ лекарственного растительного сырья и сборов в пачках и пакетах, а также фасованного «ангро»:
испытания проводят на 10 упаковках, каждую из которых взвешивают с погрешностью ± 0,01 г. Массу содержимого упаковки устанавливают как разность между массой сырья в упаковке и массой тщательно очищенной от содержимого упаковки.
Анализ лекарственного растительного сырья и сборов в фильтр-пакетах проводят по следующей методике:
10 пачек с фильтр-пакетами пробы для определения допустимых отклонений массы содержимого упаковки при промышленном фасовании вскрывают, отбирают произвольно 20 фильтр-пакетов, содержимое фильтр-пакетов высыпают и взвешивают с погрешностью ± 0,01 г. Вычисляют отклонение массы порошка в фильтр-пакете от номинальной.
Анализ лекарственного растительного сырья и сборов в брикетах проводят по следующей методике: 10 контурных ячейковых упаковок, брикетов пробы для определения допустимых отклонений при промышленном фасовании вскрывают с погрешностью ± 0,01 г. Вычисляют отклонения массы брикета от номинальной.
Допустимые отклонения массы содержимого упаковки при промышленном фасовании лекарственного растительного сырья обозначены в таблице 2.8.19.-6.
Таблица 2.8.19.-6
г-1
-
■
-j
-
Для
одной упаковки
До 100
Допустимые отклонения ± %
Для десяти упаковок 1,6
От 101 до 200 От 201 до 1000 От 1001 до 10000 Свыше 10000
3 2 1
0,2
0,9 0,6 0,3 0,06
Выборку и отбор проб из серий фасованного «ангро» лекарственного растительного сырья цельного, измельчённого и порошка проводят, как указано для лекарственного растительного сырья, исключая выделение пробы для установления степени зараженности амбарными вредителями.
В случае обнаружения живых и мёртвых вредителей в фасованной продукции лекарственного растительного сырья и сборах проводят отбор дополнительной пробы массой 500 г для их определения.
# 2.8.20. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ТОКСИЧЕСКИХ ВЕЩЕСТВ МЕТОДОМ АТОМНО-АБСОРБЦИОННОЙ СПЕКТРОСКОПИИ
Подготовка проб. Способ сухой минерализации - основан на полном разложении органических веществ путем сжигания пробы сырья в электропечи при контролируемом температурном режиме.
В кварцевый тигель берут навеску сырья массой 3 - 5 г (точная навеска). Тигель с навеской помещают в холодную муфельную печь и проводят обугливание, постепенно увеличивая температуру (на 50°С через каждые 30 минут) до 500 °С. Продолжают минерализацию до получения серой золы. Тигель вынимают из печи, охлаждают до комнатной температуры и смачивают содержимое по каплям минимальным количеством раствора 15 г/л азотной кислоты Р. Выпаривают кислоту досуха на водяной бане с последующей выдержкой в сушильном шкафу при температуре до 140 °С. После охлаждения тигель с навеской снова помещают в печь, постепенно доводят температуру до 300 °С и выдерживают в течение 0,5 ч. Указанный цикл повторяют несколько раз Минерализацию считают законченной, когда зола станет белого или слегка окрашенного цвета, без обугленных частиц. При наличии обугленных частиц повторяют обработку золы раствором азотной кислоты или водой.
Параллельно в двух тиглях проводят минерализацию добавляемых к навеске реактивов для контроля их чистоты.
Способ мокрой минерализации - основан на полном разрушении органических веществ пробы при нагревании с серной и азотной концентрированными кислотами с добавлением хлорной кислоты или перекиси водорода или при нагревании только с перекисью водорода.
Навеску сырья массой 3 - 5 г берут на беззольный фильтр, заворачивают в него и помещают на дно колбы Кьельдаля или плоскодонной колбы. В колбу вносят кислоту азотную Р из расчета 10 мл на 5 г навески и выдерживают не менее 15 мин. Можно оставить на ночь. Затем в колбу вносят 2 - 3 стеклянных шарика для равномерного кипения, закрывают грушевидной стеклянной пробкой и нагревают на электроплитке слабо, затем сильнее, упаривая содержимое колбы до объема 3 - 5 мл. Затем колбу охлаждают, вносят 10 мл кислоты азотной Р, содержимое упаривают до объема 5 мл, после чего охлаждают. Эту процедуру повторяют 2 - 4 раза. Затем в колбу вносят 10 мл кислоты азотной Р, 5 мл кислоты серной Р, 4 мл кислоты хлорной Р (или вместе кислоты хлорной Р 4 мл концентрированного раствора пероксида водорода Р) из расчета на 5 г сырья. Не допускается изменять последовательность внесения кислот, хлорная кислота всегда вносится последней. Содержимое колбы упаривают до объема около 5 мл, не допуская образования коричневой окраски жидкости. При появлении коричневой окраски нагревание прекращают.
Колбу охлаждают до комнатной температуры, добавляют 5 мл кислоты азотной Р и 2 мл кислоты хлорной Р (или вместо кислоты хлорной Р 2 мл концентрированного раствора пероксида водорода Р) и снова нагревают до появления белых паров серного ангидрида. Если при этом раствор не обесцветился, эту процедуру повторяют. Минерализацию считают законченной, если раствор после охлаждения остается бесцветным или бледно-желтым.
Для удаления остатков кислот в охлажденную колбу добавляют 10 мл воды Р и кипятят 10 мин с момента выделения белых паров, затем охлаждают. Добавление воды Р и нагревание повторяют еще 2 раза. Если при этом образуется осадок, в колбу вносят 20 мл воды Р, 2 мл кислоты серной Р, 5 мл кислоты хлористоводородной Р и кипятят до растворения осадка, постоянно дополняя испаряющуюся воду.
Параллельно проводят минерализацию добавляемых реактивов для контроля их чистоты.
Золу, полученную способом сухой минерализации, растворяют в тигле при нагревании в кислоте азотной разведённой Р из расчета 1 - 5 мл кислоты на навеску е зависимости от зольности сырья. Раствор выпаривают до влажных солей. Осадок растворяют в 15 - 20 мл раствора 15 г/л азотной кислоты Р, количественно переносят в мерную колбу вместимостью 25 мл и доводят до метки той же кислотой.
При неполном растворении золы полученный раствор с осадком упаривают до влажных солей, перерастворяют в минимальном объеме смеси воды Р и хлористоводородной кислоты Р (1:1) по объему, еще раз упаривают до влажных солей и растворяют в 15 - 20 мл раствора 28 г/л хлористоводородной кислоты Р. Раствор количественно переносят в мерную колбу вместимостью 25 мл и доводят до метки той же кислотой.
При неполном растворении золы полученный раствор с осадком доводят до объема 30 - 40 мл раствором 28 г/л хлористоводородной кислотой Р и подогревают на водяной бане или электроплитке при слабом нагреве в течение 0,5 ч. Если в этом случае полного растворения не наблюдается, раствор отфильтровывают через промытый растворителем фильтр, осадок промывают и отбрасывают, а фильтрат переносят в мерную колбу вместимостью 50 мл и доводят до метки той же кислотой.
При использовании способа мокрой минерализации полученный раствор минерализата упаривают до влажных солей и продолжают растворение по указанной схеме.
Определение токсических элементов проводят методом атомно-абсорбционной спектроскопии согласно 2.2.23.
Подготовка проб для определения содержания мышьяка. В чашу с точной навеской сырья (3 - 5 г) добавляют 10 % от массы навески магния оксида Р и такое же количество раствора магния нитрата Р в спирте Р (в расчете на безводную соль) Полученную смесь тщательно перемешивают до образования однородной кашицы.
Допускается минерализация сырья без добавления смеси магния оксида Р и магния нитрата Р.
Чашу помещают на водяную баню или в сушильный шкаф при температуре 80 - 10С °С и выпаривают досуха, после чего переносят на электроплитку и обугливают при слабом нагреве до прекращения выделения дыма. Затем чашу помещают в электропечь, ранее отрегулированную на температуру 250 °С, повышают температуру до 450 °С постепенно на 50 °С в час и продолжают минерализовать в этих условиях до получения серой золы. Далее проводят минерализацию. Чашу вынимают из печи, охлаждают до комнатной температуры и смачивают содержимое по каплям минимальным количеством воды Р. Выпаривают воду досуха на водяной бане с последующей выдержкой в сушильном шкафу при температуре до 140 °С. После охлаждения чашу с навеской снова помещают в печь, постепенно доводят температуру до 300 °С и выдерживают в течение 0,5 ч. Указанный цикл повторяют несколько раз. Минерализацию считают законченной, когда зола станет белого или слегка окрашенного цвета, без обугленных частиц. При наличии обугленных частиц повторяют обработку золы водой Р.
Параллельно в двух чашках проводят минерализацию добавляемых к навеске реактивов для контроля их чистоты.
Далее определение содержания мышьяка проводят согласно 2.4.2.
# 2.8.21. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ РАДИОНУКЛИДОВ
Требования настоящей статьи применяются в отношении лекарственного растительного сырья независимо от формы выпуска на этапах разработки и постановки на производство новых видов лекарственных средств при его переработке, производстве, хранении, транспортировке, закупке, ввозе в страну, сертификации и реализации (далее обращение лекарственного растительного сырья).
Государственному контролю на радиационную безопасность подлежит лекарственное растительное сырьё, выпускаемое предприятиями различных форм собственности на территории Республики Беларусь и ввозимое на территорию Республики Беларусь.
Термины и определения
В настоящей фармакопейной статье применяются следующие термины и определения:
Счётный образец - аналитическая проба - определённое количество пробы выделенной методом квартования из объединённой пробы для измерений его радиационных параметров.
Радиоактивность - испускание ионизирующих излучений при самопроизвольном превращении радиоактивных ядер.
Активность радионуклида - отношение числа dN самопроизвольных превращений ядер данного радионуклида, происходящих за интервал времени dt, к этому интервалу.
, dN
А =
dt
Единица активности - Беккерель (Бк) - одно ядерное превращение в секунду.
Удельная активность радионуклида (q) - отношение активности радионуклида е исследуемом образце к массе (объёму) исследуемой пробы (Бк/кг, Бк/л).
Q=М
М
Концентрирование удельной активности - процедура приготовления счётного образца путём высушивания, обугливания, озоления или химического концентрирования.
Средства измерения - включают в себя необходимые для определения удельной активности радионуклидов цезия-137:
радиометрическая установка с приспособлениями для экспонирования счётных образцов;
методики выполнения измерений на данной радиометрической установке;
методики приготовления счётных образцов вместе с необходимыми устройствами, приспособлениями и инструментами.
Радиационный контроль - применение средств измерений для определения соответствия исследуемых образцов требованиям нормативов норм радиационной безопасности.
Общие положения
Настоящая фармакопейная статья рассматривает вопросы радиационного контроля лекарственного растительного сырья, полуфабрикатов, потребительских упаковок и сборов, применяемых в сфере обращения лекарственных средств.
При проведении радиационного контроля лекарственных средств в сфере обращения лекарственных средств, выполняются следующие стандартные процедуры:
отбор проб из партии лекарственного растительного сырья или от серии лекарственных средств;
приготовление счётных образцов, с концентрированием удельной активности в случае необходимости;
измерение активности цезия-137 в счётных образцах;
расчёт результатов измерений и погрешностей исследований;
определение соответствия лекарственных средств критериям радиационной безопасности.
Отбор проб для радиационного контроля проводится согласно 2.8.19. Подготовка счётных образцов лекарственных средств для определения удельной активности цезия-137 проводится на основании настоящей фармакопейной статьи.
Персонал, осуществляющий радиационный контроль лекарственных средств, должен пройти соответствующее обучение для работы на радиометрических установках, освоить методики выполнения измерений и методики приготовления счётных образцов.
Результатом измерений активности радионуклидов в счётном образце А является интервал значений активности от (А - ДА) до (А + ДА), в котором с вероятностью Р=0,95 находится значение измеряемой величины А (Бк).
А - ДА < А < А + ДА (1)
где:
А - значение измеренной активности радионуклида на данной радиометрической установке (Бк).
ДА - значение погрешности при измерении А на данной радиометрической установке.
Пример. При измерении активности радионуклида получены следующие значения: А = 75 Бк; ДА = 20 Бк.
75 - 20 < 75 < 75 + 20 — 55 < 75 < 95 с вероятностью Р = 0,95 значение измеряемой величины А (Бк) находится в интервале от 55 до 95 Бк.
Если часть интервала от (А - ДА) до А, или {(А - ДА) - А} находится в области отрицательных значений, то следует считать, что с вероятностью Р = 0,95 значение величины А находится в интервале:
От 0 до (А + ДА) или {0 - (А + ДА)} (2) Пример. При измерении активности радионуклида получены следующие значения: А = 15 Бк; ДА = 20 Бк.
15 - 20 < 15 < 15 + 20 (1) — 0 < 15 < 35 (2) с вероятностью Р = 0,95 значение измеряемой величины А (Бк) находится в
интервале от 0 Бк до 35 Бк.
Если значение активности А < 0, то следует считать, что значение величины А
находится в интервале:
От 0 до ДА или {0 - ДА} (3) Пример. При измерении активности радионуклида получены следующие значения:
А = -5 Бк; ДА = 20 Бк.
Т.к. А < 0 -5 < Г~- 0 - 20 (3) с вероятностью Р = 0,95 значение измеряемой величины А (Бк) находится в интервале от 0 Бк до 20 Бк
Результатом определения удельной активности радионуклидов в лекарственном растительном сырье Q (Бк/кг), является интервал, характеризуемых соотношением:
Q - ДQ < Q < Q + ДQ
где:
Q - значение удельной активности радионуклида в исследуемом лекарственном растительном сырье;
ДQ - значение погрешности.
Порядок отбора проб лекарственного растительного сырья.
Порядок отбора проб лекарственного растительного сырья включает выделение однородной по радиационному составу пробы для приготовления счётных образцов. Отбор проб должен при оптимальных затратах времени и средств обеспечить представительность лекарственных средств, находящихся на любом этапе обращения. Отбор проб осуществляется согласно 2.8.19.
Примечание. Перед отбором точечных проб от выбранных транспортных единиц сырья, целесообразно выполнить дозиметрический контроль по мощности дозы гамма-излучения для определения безопасности партии сырья с помощью поисковых радиометров типа ДРГ-01Т, ДКС-96П, ДКС-96К, ДКС-96М, ДКС-96Б, СРП-88Н, МКС-04Н, ДКГ-02У. Определение однородности партии сырья возможно с помощью радиометрической установки, в этом случае анализируются точечные пробы из транспортных единиц сырья до составления объединённой пробы. Если в результате предварительного дозиметрического или радиационного контроля партии сырья установлена неоднородность партии или превышение допустимых показателей, то этот факт должен быть отмечен в акте отбора объединённой пробы. Решение о приёмке или об отказе от приёмки сырья может быть принято только после проведения полного анализа в соответствии с данными методическими указаниями.
Для измерения удельной активности цезия-137 в лекарственном растительном сырье и определения его соответствия критериям радиационной безопасности при оптимальных затратах времени и средств предлагается три варианта подготовки счётных образцов. В таблице 2.8.21.-1 приведены ориентировочные массы в зависимости от используемого варианта измерений.
Таблица
2.8.21.-1
Массы
навесок (не менее), г; определение
Cs-137
3
вариант измерений
|
1 |
Корни алтея |
40 |
280 |
400 |
|
2 |
Плоды аниса |
40 |
400 |
600 |
|
3 |
Корневища аира |
45 |
300 |
600 |
|
4 |
Почки берёзы |
50 |
450 |
600 |
|
5 |
Цветки бессмертника |
15 |
150 |
250 |
|
6 |
Плоды боярышника |
50 |
350 |
600 |
|
7 |
Листья брусники |
30 |
170 |
350 |
|
8 |
Побеги багульника |
30 |
225 |
350 |
|
9 |
Корневища с корнями валерианы |
40 |
300 |
450 |
|
10 |
Корневища и корни девясила |
50 |
350 |
650 |
|
11 |
Трава донника |
25 |
150 |
300 |
|
12 |
Кора дуба |
50 |
300 |
500 |
|
13 |
Трака душицы |
25 |
200 |
300 |
|
14 |
Трава зверобоя |
30 |
170 |
350 |
|
15 |
Кора крушины |
25 |
300 |
400 |
|
16 |
Столбики с рыльцами кукурузы |
25 |
150 |
250 |
|
17 |
Цветки липы |
30 |
200 |
450 |
|
18 |
Корни лопуха |
40 |
350 |
500 |
|
19 |
Семена льна |
60 |
600 |
800 |
|
20 |
Листья мать-и-мачехи |
30 |
200 |
350 |
|
21 |
Трава мелиссы |
25 |
180 |
250 |
|
22 |
Плоды можжевельника |
50 |
400 |
600 |
|
23 |
Слоевища ламинарии |
40 |
600 |
800 |
|
24 |
Листья мяты |
25 |
180 |
250 |
|
25 |
Цветки ноготков |
16 |
180 |
270 |
|
26 |
Листья ортосифона (почечный чай) |
30 |
260 |
400 |
|
|
|
|
| |
|
27 |
Цветки пижмы |
25 |
220 |
350 |
|
28 |
Листья подорожника |
35 |
280 |
400 |
|
29 |
Трава пустырника |
20 |
180 |
300 |
|
30 |
Плоды расторопши |
60 |
600 |
800 |
|
31 |
Корневища и корни родиолы |
30 |
200 |
400 |
|
32 |
Плоды рябины |
60 |
370 |
600 |
|
33 |
Листья сены |
40 |
180 |
250 |
|
34 |
Трава спорыша |
25 |
220 |
350 |
|
35 |
Корни солодки |
30 |
270 |
400 |
|
36 |
Почки сосны |
25 |
150 |
300 |
|
37 |
Плоды тмина |
45 |
300 |
500 |
|
38 |
Листья толокнянки |
35 |
200 |
450 |
|
39 |
Трава тысячелистника |
15 |
200 |
300 |
|
40 |
Плоды укропа |
40 |
330 |
500 |
|
41 |
Створки фасоли |
20 |
180 |
280 |
|
42 |
Плоды фенхеля |
40 |
380 |
500 |
|
43 |
Трава фиалки |
25 |
200 |
300 |
|
44 |
Трава хвоща |
25 |
130 |
280 |
|
45 |
Соплодия хмеля |
20 |
190 |
300 |
|
46 |
Трава чабреца |
30 |
170 |
300 |
|
47 |
Чага |
25 |
350 |
400 |
|
48 |
Побеги черники |
25 |
150 |
280 |
|
49 |
Трава череды |
25 |
200 |
300 |
|
50 |
Трава чистотела |
30 |
150 |
300 |
|
51 |
Листья шалфея |
35 |
250 |
350 |
|
52 |
Плоды шиповника |
50 |
400 |
600 |
|
53 |
Листья эвкалипта |
30 |
240 |
400 |
|
54 |
Корневища и корни элеутерококка |
40 |
400 |
600 |
|
55 |
Трава эрвы шерстистой |
25 |
200 |
350 |
|
Таблица 2.8.21 .-2 | ||||
|
№ 1 2 3 4 5 6 7 8 9 10 |
Наименование |
Массы навесок (не менее), г; определение Cs-137 | ||
|
1 вариант измерений |
2 вариант измерений |
3 вариант измерений | ||
|
Побеги |
30 |
200 |
340 | |
|
Почки |
40 |
300 |
450 | |
|
Цветки |
25 |
200 |
300 | |
|
Плоды |
50 |
400 |
600 | |
|
Листья |
30 |
200 |
320 | |
|
Трава |
30 |
200 |
300 | |
|
Кора |
35 |
250 |
400 | |
|
Семена |
60 |
600 |
800 | |
|
Корни |
40 |
330 |
500 | |
|
Корневища |
40 |
360 |
600 | |
|
11 |
Корневища с корнями |
40 |
300 |
450 |
Определение цезия-137 при экспонировании счётного образца 1800 сек (30 мин).
вариант измерений - предполагает использование аттестованной геометрии -Denta 100 мл, измельчение указанной массы до фракции «крупный порошок», проходящей сквозь сито с отверстиями размером 2 мм (фракция из фильтр-пакетов может использоваться без дополнительного измельчения).
вариант измерений - предполагает использование аттестованной геометрии -сосуда Маринелли объёмом 1 л, сырьё фракции «измельчённое» (проходящее через сито 8 мм; сырьё, фасованное в пачку), допускается без измельчения анализировать цельное сырьё, фасованное в пачку, например, плоды боярышника, плоды шиповника, семена льна и ит.д., при достаточной насыпной массе.
вариант измерений - предполагает использование аттестованной геометрии -сосуда Маринелли объёмом 1 литр, измельчение указанной массы до фракции «крупный порошок», проходящей сквозь сито с отверстиями размером 2 мм.
Приготовление счётных образцов и измерение активности цезия-137 в пробах лекарственного растительного сырья.
Подготовка проб к измерениям. Подготовка проб к измерениям включает в себя отбор пробы, предварительное измельчение сырья с целью приготовления однородного счётного образца в соответствии с выбранным вариантом измерения, объёмом её заполнения и схемой анализа:
пробу лекарственного растительного сырья предварительно измельчают с помощью ножа, секатора, мясорубки, тёрки, а затем в специальных лабораторных измельчителях, дробилках и мельницах, возможно использование кофемолки.
в зависимости от выбранного варианта измерения, радиологическому исследованию подвергается лекарственное растительное сырьё со степенью измельчения 8 мм, 2 мм и 1 мм, т.е. частицы, проходящие сквозь сито с отверстиями размером 7 мм, 2 мм и 1 мм.
масса навески для радиологического исследования определяется для каждого вида сырья и выбора аттестованной геометрии и не может быть меньше указанной в таблице 2.8.21.-1.
Приготовление счётного образца для измерения цезия-137 зависит от поступившего на анализ сырья, используемого метода измерения и чувствительности используемой радиометрической установки. Сырьё, фасованное в пачку, как правило, измельчённое и проходит сквозь сито с отверстиями размером 8 мм. Сырьё, фасованное в фильтр-пакеты, как правило, «крупный порошок» и проходит сквозь сито с отверстиями размером 2 мм.
Брикет после разрушения представляет собой порошок, который, как правило, проходит сквозь сито с отверстиями размером 1 мм. Выбор аттестованных геометрий (сосуд Маринелли 1 л и Denta 100 мл) зависит от результатов, полученных в процессе анализа.
Приготовление счётного образца для определения удельной активности цезия-137. Пробу измельчают и просеивают сквозь сито с отверстиями 2 мм для аттестованной геометрии Denta 100 мл (1 вариант измерений), 7 мм (2 вариант измерений) или 2 мм (3 вариант измерений) для сосуда Маринелли. Измельчённое лекарственное растительное сырьё помещают в сосуд Ма.ринелли или Denta 100 мл, масса сырья не может быть меньше указанной в таблице 2.8.21.-1, и измеряют активность цезия-137.
Для определения массы измеряемого счётного образца сосуд взвешивают до и после его заполнения.
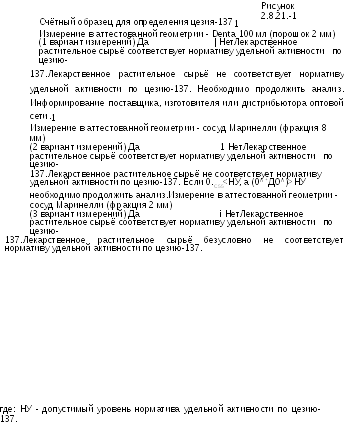
При получении отрицательного результата в Denta 100 мл (т.е. сырьё относится ко второй или третей группе радиационной безопасности) увеличивают массу счётного образца (2 вариант измерений) и повторно проводят измерение активности цезия-137 в сосуде Маринелли. При получении третей группы по критериям радиационной безопасности продолжают исследование по 3 варианту измерений. Схема проведения радиационного контроля приведена на рисунке 2.8.21.-1.
Измерение удельной активности цезия-137. Для измерения удельной активности цезия-137 рекомендуется использовать гамма-спектрометры со сцинтилляционными и полупроводниковыми детекторами, находящимися в свинцовой защите, толщина которой должна быть не менее 50 мм. Минимальная измеряемая активность подобных гамма-спектрометров 3-10 Бк.
Установленная настоящей статьёй масса анализируемой пробы в зависимости от выбранного варианта измерений обеспечивает приемлемую погрешность получаемого результата при измерении в стандартной геометрии - Denta 100 мл и сосуд Маринелли (1
л).
Первоначально измерение активности проводят в аттестованной геометрии - Denta 100 мл; как правило по первому варианту измерений чувствительности гамма-спектрометра не хватает для получения достоверного результата - увеличивают массу счётного образца и в качестве измерительного сосуда используют сосуд Маринелли (2 или 3 вариант измерений).
Измерение активности производится в соответствии с инструкцией к используемому гамма-спектрометру и настоящей статьёй. Рекомендуется использование компьютеризированных гамма-спектрометрических комплексов с программным обеспечением.
Обработка (вычисление) результатов измерения осуществляется в зависимости от типа использованного гамма-спектрометра.
Схема проведение радиационного контроля приведена на рисунке 2.8.21.-1.
В схеме под утвердительном ответом «Да» подразумевается соответствие полученного результата нормативу удельной активности цезия-137 в лекарственном растительном сырье. Ответ «Нет» - если сырье относится ко второй или третьей группе (по критериям радиационной безопасности).