

1.6. Модели идеальных систем в химической термодинамике. Реальные системы
Построение модельных представлений является одним из важнейших элементов научного познания. Это обусловлено тем, что понимание особенностей поведения реальных систем начинается с момента, когда удается предсказать, какими свойствами обладала бы рассматриваемая система в "идеальном " случае.
В химической термодинамике особое значение имеют две модели идеальных систем, а именно: модель идеального газа и модель идеального раствора. Каждая из них может быть строго определена. Однако на начальном этапе достаточно ограничиться качественным рассмотрением этих моделей с единственной целью - понять значение и главные принципы, положенные в основу моделирования указанных систем.
Модель идеального газа, подчиняющегося уравнению Клапейрона-Менделеева*-*:
*' Химики в России обычно называют его уравнением Менделеева-Клапейрона, оставляя без внимания тот факт, что уравнение pV = ВТ, где В - константа, зависящая от массы газа и его
молярной массы, было предложено Б.П.Э. Клапейроном в 1834 г, т.е. в год рождения Д.И. Менделеева. В отличие от химиков, физики называют это уравнение либо уравнением Клапейрона, либо Клапейрона-Менделеева, но не наоборот, что вполне справедливо.
[p# 23]
22
[p# 24]
PV = nRT,
где n - число молей газа; R - универсальная газовая постоянная [Дж/(моль К)], исходит из того, что газ состоит из точечных материальных частиц конечной массы, между которыми отсутствуют силы, действующие на расстоянии и которые сталкиваются между собой по законам соударений упругих шаров. Основываясь на этих представлениях, стало возможным объяснить причины отклонений свойств реальных газов от поведения идеальных. Эти причины связаны с существованием сил межмолекулярного взаимодействия и наличием у молекул собственного объема.
Концепция идеального газового раствора также основывается на допущении, что система состоит из невзаимодействующих между собой частиц, при смешивании которых каждый газ ведет себя по отношению к другому как пустота. Естественными следствиями этого являются законы Дальтона (1805) и Амага(1880).
Закон Дальтона утвержает, что парциальное давление компонента газовой смеси (р$ равно произведению его
мольной доли (х$ на общее давление (р):
Pi =XiP,
где xj - мольная доля, равная отношению числа молей данного компонента смеси к общему числу молей:
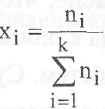
Согласно закону Амага:
объем газовой смеси, при фиксированной температуре и давлении является аддитивной функцией ее состава:
![]()
где V - общий объем смеси, nk иУ£ - число молей и объем одного моля k-ro компонента при давлении, равном общему давлению газовой смеси.
Кажется очевидным, что в конденсированных фазах, т.е. в случае жидких и твердых растворов, пренебрегать межмолекулярным взаимодействием не
1. Терминология
[p# 25]
представляется возможным из-за малости расстояния между частицами. В этой ситуации, модель идеального раствора модифицируют, допуская отсутствие специфического взаимодействия между разноименными компонентами. Иными словами, как и прежде, исходят из предположения, что при смешивании разнородных частиц они как бы не замечают присутствия друг друга вследствие того, что энергии взаимодействия одноименных и разноименных молекул оказываются одинаковыми:
Е1-2 =Е1-1 = Е2-2
По этой причине свойства идеального раствора не зависят от природы растворенных веществ, а определяются только их концентрациями. Согласно закону Рауля (1886 г.),
если один или все компоненты раствора являются летучими, то парг^иальное давление пара каждого из них над раствором при заданной температуре пропорционально доле молекул данного компонента в растворе:
![]()
где р? - давление насыщенного пара над чистым компонентом.
При сделанных выше ограничениях справедливость закона аддитивности объема раствора воспринимается уже как самоочевидный факт.
Учитывая то обстоятельство, что реальные системы даже в случае газов, лишь приближаются к идеальным, ясно, что в практическом отношении очень важно определить принципы подхода к описанию отклонений свойств реальных объектов от поведения идеальных систем. Существуют два подхода к решению этой задачи. В первом - пытаются количественно охарактеризовать энергетику межмолекулярных взаимодействий, приводящих к отклонениям от идеальности. С этой целью рассматривают парные, тройные и более сложные взаимодействия с помощью функций потенциальной энергии. В настоящее время этот подход не получил распространения из-за значительных трудностей вычислительного характера. Альтернативой ему является предложенное Льюисом преобразование, смысл которого сводится к чисто формальной характеристике отклонений от идеальности с помощью экспериментально определяемых поправочных коэффициентов, которые и являются мерой отклонения свойств реальных систем от идеальных.
В случае реальных газов вводится понятие эффективного давления или летучести (f), которая отличается от давления идеального газа при заданных
24
[p# 25]
V и Т на величину поправочного коэффициента, называемого коэффициентом летучести (у), и определимого из уравнения:
![]()
Практическое удобство преобразования, предложенного Льюисом, заключается в том, что использование функции летучести исключает необходимость поиска новых уравнений, характеризующих поведение реальных систем. Замена давления на летучесть позволяет применять для реальных систем все уравнения, которые установлены для идеальных систем
В случае растворов вводится понятие эффективной концентрации или активности (а^, которая связана с концентрацией уравнением:
![]()
где Xj - мольная доля, -^ - коэффициент активности.
Подход Льюиса не позволяет охарактеризовать в количественной форме природу сил, приводящих к отклонениям от идеальности, однако он вполне строго характеризует эти отклонения.
Используя понятия летучести и активности, представляется возможным найти значения всех термодинамических величин, определяющих поведение реальных систем. Поэтому неудивительно, что на современном этапе подход Льюиса играет доминирующую роль при изложении большинства термодинамических разделов курса физической химии
1. Терминология 25
[p# 26]