

63.Температурная зависимость наблюдаемой скорости реакции
Большое влияние на хар-р протекания гетер.х.р. оказ-ют давления реагир.в-в,скор. потоков,пористость К и тем-ра.При изменении тем-ры на 10ºС скор. диф-зии измен-ся ≈в 1,2 раза, а скор.х.р.- в 3-4раза→при понижении тем-ры скор.х.р. убывает быстрее,чем скор. диф-зии,и при низких тем-рах пр-с чаще протекает в кинетич.обл.Если не все участки пов-ти, на к-рой протекает р-я в равной степени доступны для реагир.м-л,то часто оказ-ся,что на части пов-ти р-я протекает в кинетич.обл,а на остальной-в диф-ной.
Гетерогенная хим. р-ция может протекать в том случае, если происходит непрерывная молекулярная или конвективная диффузия реагирующих в-в к поверх-ти, на кот. идет данная р-ция, и непрерывная обратная диф-зия продуктов р-ции.Скор. пр-са будет опр-ся скор. лимит-щей стадии.Если скор. хим. р-ции > скор. диф-зии, то скорость пр-сса в целом будет опред-ся скор-тью диф-зии. В этом случае пр-с лежит в диф-оной области и кинетика р-ции подчиняется урав-ям, к-рые рассматр-ют пр-сы диф-зии.Пр-с при этом чаще всего опис-ся ур-ем 1-го порядка,т.к. скор.диф-зии прямо пропорц-на конц. Если скор. х. р. < скор. диф-зии, то скорость р-ции опр-ся скоростью хим. р-ции. Тогда пр-с лежит в кинетической области и описывается урав-нием кинетики той р-ции, к-рая проходит на повер-ти К. Если скорось диф. и скорость хим. р-ции, рассмотренные независимо друг от друга, соизмеримы, то имеется переходная область.Она хар-ся тем,что конц. реагир.в-в в центре зерна меньше,чем на наруж пов-ти зёрен К.В перех.обл. порядок и Еа измен-ся от измен. тем-ры,к-рое происходит от тем-ры, опр-мой кинетич.обл.,до тем-ры,хар-ной для диф.обл.Большое влияние на хар-р протекания гетер.х.р. оказ-ют давления реагир.в-в,скор. потоков,пористость К и тем-ра.При изменении тем-ры на 10ºС скор. диф-зии измен-ся ≈в 1,2 раза, а скор.х.р.- в 3-4раза→при понижении тем-ры скор.х.р. убывает быстрее,чем скор. диф-зии,и при низких тем-рах пр-с чаще протекает в кинетич.обл.Если не все участки пов-ти, на к-рой протекает р-я в равной степени доступны для реагир.м-л,то часто оказ-ся,что на части пов-ти р-я протекает в кинетич.обл,а на остальной-в диф-ной.При проведении р-ции на неравнодоступ.пов-ти возмож.4 обл.1)Внеш.диф.обл. (Скор.диф. м-л к внеш. пов-ти К-лимит.ст).Внешдиф.обл. проявл-ся,когда пр-с проводится при повыш. тем-рах,при наличии у пов-ти К плёнки,затруд-щей подход м-л реаг-тов к актив.ц. К,при проведении пр-са в присут-вии высокоактив.К. В случ. внеш.диф. конц-ии регаир.в-в на пов-ти пористого тела и внутри пор гораздо меньше,чем в объёме.2)Внутр.диф.При таком режиме конц. реагир. в-в у внеш.пов-ти пористого К близки к конц. их в объёме;конц в порах уменьш-ся от наруж. пов-ти зёрен пористого К к их центру,а конц. прод.р-ции соот-но возрастает.Кин-ка пр-са завис. от соотнош. внеш и внутр пов-ти;если они соизмеримы,то кин-ка соот-ет промеж.обл.Внутридиф.пр-сы опис-ся ур-ем массопереноса: wм.п.=β∙S(cп-соб),β=D/l-коэф-т массопер-са; cп-конц в-ва у пов-ти,на входе в пору,соб-конц в-ва в объёме пор.3)Внеш.кинетич.обл.(Скор. х.р. м\у адсорбир.в-ми на внеш. пов-ти К гораздо меньше скор. пр-сов на др. стадиях). Конц. реагир. в-в на пов-ти К и в объёме равны,но р-я протекает т-ко на внеш пов-ти К.Кин-ка пр-са опр-ся кин-кой х.р.Состав-ся ур-я завис-ти скор.х.р. от тем-ры, конц К и др.параметров.4)Внутр.кинетич.обл.(наиб медл.стадия-ст. хим. превращ-я м-л реагентов,адсорбир-ных на актив.ц. внутри пор К).При таком режиме конц в-в в объёме,на пов-ти К и внутри пор одинаковы.Кин-ка опр-ся к-кой пр-са.Скор.пр-са зависит от всей пов-ти К(внутр. и внеш).А во внешнекин.обл. т-ко от внеш.
64.З-ны Фика и роль диффузии в гетерог.катал.р-циях.
Гетероген.пр-с можно разделить на 5 стадий:1)транспорт реагир.в-в к пов-ти К(диф-зия);2)адс-ция реагир.в-в на пов-ти К;3)р-ция на пов-ти К;4)десорбция продуктов р-ции с освобождением пов-ти К;5)транспорт продуктов р-ции в объёме(диф-зия).В завис-ти от условий провед-я пр-са и его особ-тей наиб.медленной стадией может быть любая из 5.Для гетероген.пр-сов особую важность приобретает перемещение в-ва из внутрен.объёма ж-ти или газа к тв.пов-ти,т.к. гетероген. хим. р-ция может протекать в том случае, если происходит непрерывная молекулярная или конвективная диффузия реагирующих в-в к поверх-ти, на кот. идет данная р-ция, и непрерывная диф-зия продуктов р-ции.Диф-ный пр-с имеет чисто физич.природу и его можно описать с пом. з-нов Фика:dn/dt=-D*S*(dc/dl) (1); dn/dt=-D*S*(d2c/dl2) (2),n-кол-во в-ва,проход-го ч\з площадку размером S за вр t;D-коэф-т мол-ной диф-зии,м2*с-1.1-ый з.Фика опр-ет,что скор. диф. пр-са в неподвижной среде в изотермич.условиях ч\з слой толщиной δ прямо пропорц-на градиенту конц-ции в-ва.Отриц.знак в (1) показ-ет убыль конц-ции;коэф-т D при малых конц-ях не завис. от конц.в-ва С. 2-ой з.Фика опр-ет скор.накопления в-ва в дан. системе за опред-ное вр.Коэф-т диф-зии опр-ся как отнош. квадрата средней длины пробега м-л ко вр.диф-зии и он обратно пропорционален давлению: D=l2сред/2t.Градиент конц. в-в в потоке можно представить в форме конечной разности: -dn/dl=(cs-cx)/l (3).Подставим (3) в (1): dn/dt=D*S*((cs-cx)/l); cs,cx- конц в-ва в объёме и у пов-ти К,моль\м3.Скорость диф-зии возрастает с повыш.тем-ры по з-ну,аналог.ур.Аррениуса: D=ke-E/RT.Но величина Е во много раз меньше Еа большин-ва х.р.→с повыш тем-ры скор.диф-зии будет расти значит-но медленнее,чем скорость х.р.
66.Кинетика гетерогенных каталитических реакций, проводимых в реакторе идеального вытеснения.
Реакторы проточного типа могут применяться двух типов: пустотелые или имеющие распределительные решетки. Процессы в таких реакторах проходят в потоке, в реакторе идеального вытеснения реакционной смеси с неподвижным слоем катализатора или в потоке. В этом случае через слой неподвижного шарикового или движущегося с малой скоростью катализатора сырье проходит в поршневом режима, то есть, когда предыдущий слой сырья вытесняет передний его фронт.
Скорость гетерогенной каталитической для режима идеального вытеснения
Гидродинамический РИВ реакционной смеси может соблюдаться для реактора, в котором отношение длины его к диаметру больше пятидесяти (l / d > 50). Уравнения скорости реакций, протекающей в потоке, можно получит на основе анализа уравнения потока массы веществ.
Гомогенный поток массы веществ в нестационарных условиях, находящийся под воздействием конвективной силы к в котором проходит химическая реакция:
![]() (1),где
(1),где
![]() - объемно-молекулярная концентрация,
NA, VA-мольная и объемная скорости подача
веществ,V - линейная скорость перемещения
потока веществ,
- объемно-молекулярная концентрация,
NA, VA-мольная и объемная скорости подача
веществ,V - линейная скорость перемещения
потока веществ,
w - скорость химической реакции в гомогенной системе, τ-время.
Для перехода к уравнению потока массы веществ через слой зернистого катализатора необходимо учитывать тот факт, что вещество перемещается только между зернами катализатора.
Вводим
коэффициент
![]() ,
определяющий долю площади реактора,
свободной от катализатора.
,
определяющий долю площади реактора,
свободной от катализатора.
S0V-площадь
единицы объема катализатора. Умножим
уравнение (1) на
![]() /S0V
/S0V
![]()
Введем
обозначение![]()
Запашем
концентрацию и лилейную скорость через
соотношения:
![]() ;
V=Va/Ps*χ. Ps-площадь раствора в сечении
Получим:
;
V=Va/Ps*χ. Ps-площадь раствора в сечении
Получим:
![]()
Для
стащюиарных условий (для равномерной
подачи сырья в реактор) концентрация
вещества А не меняется со временем по
длине реактора И тогда первая производная
по времени будет равна нулю
![]()
Уравнение
потока массы веществ для стационарных
условий:
![]()
Уравнение
скорости химической реакции, протекающей
в потоке, в гетерогенной, системе, для
РИВ. Если количество непрореагировавшего
вещества А обозначить как
![]() ,
тогда
,
тогда
![]() n0а –мольн.скорость подачи А.
n0а –мольн.скорость подачи А.
х-превращение, моль/моль. Это ураанв&ао скорости было выведено впервые АД,Баландиным и повторено с теоретическим выводом Г.М.Дакчевкоаым,
Скорость
гетерогенной каталитической реакции
зависит от количества сорбированного
катализатором вещества, от давления,
темперы, природы катализатора, наличия
в реакционной смеси астехиометрических
компонентов и других факторов. Скорость
гетерогенной каталитической реакции
будет прямо пропорциональна поверхностной
концентрации реагирующих веществ,
возведенных в степень соответствующих
порядку реакции. Для
![]() Уравнение кинетики.:
Уравнение кинетики.:
![]()
П-знак произведения., θ-относительные поверхностные концентрации веществ.
67.Связь м\у истинной и кажущ-ся энериями активации.
Если
построить на основании экспер-ных
данных график завис-ти кажущ-ся константы
скорости k* от 1/T,то получ-ся прямая,опис-мая
ур-ем:lnk*=-E*/RT+lnc или dlnk*/dt=E*/RT2(1),где
E*-кажущ-ся Еа.Найдём завис-ть м\у Еа и
E*. k*=kK.Адсорбцион.коэф-т(константа
рав-сия) К равен отнош. констант скоростей
пр-сов адс-ции и десорб-ции:К=k1/k2, k*=k∙
k1/k2(2).Каждая из констант,входящих в
(2),явл-ся функцией тем-ры.Т.к.
k1=C’/√T(C’-величина постоянная),
k2=C”e-λ/RT(3)(C”-велич.постоян.,λ-т-та
адсорбции),k=Ce-E/RT(4).Константа скорости
пр-са адс-ции k1 с тем-рой измен-ся очень
мало по срав-ю с k и k2.Завис-тью k1 от
тем-ры можно пренебречь и считать,что
эта величина постоянная.Подставив (3)
и (4) в (2):k*=Ce-E/RT∙k1/(C’e-λ/RT)→lnk*=-E/RT+λ/RT+lnconst
(const=Ck1/C’). Дифференц-ем по
Т:dlnk*/dT=(E-λ)/RT2(5)/Сравнивая (5) и (1),видим:
Е*=Е-λ(6).Это выр-е хар-ет завис-ть кажущ-ся
Еа от истин. Е* меньше Еа на величину
т-ты адс-ции реагир.в-ва.Для простешей
р-ции А→В на пов-ти тв.К можно выделить
с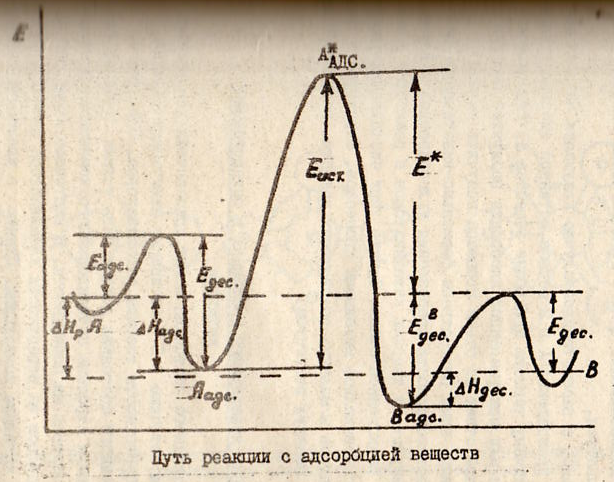 тадии:А→Аадс→А*адс
→Вадс→В. Вначале м-лы в-ва А адс-ся на
пов-ти тв.К,преодолевая энерг.барьер
Еадс.Выделяя избыток энергии в форме
т-ты адс-ции,м-ла может перейти в более
уст-вое адсор-ное сост.Аадс.На новом
кат-ком центре или под воздействием
актив.центра К м-ла А может перейти в
электронно-возбуждённое сост с переходом
на энергетический барьер высотою
Еист.(А*адс).Возбуждённые м-лы А,скатываясь
в ложбину продукта р-ции,превращсяв
прод.р-ции.При этом м-ла В переходит в
адсорбир.сост.Для десорбции м-лы В с
актив.ц. ей необх-мо преодолеть энергию
ЕВадс.После чего м-ла В может уйти с
каталит.центра в объём реакцион.смеси.
тадии:А→Аадс→А*адс
→Вадс→В. Вначале м-лы в-ва А адс-ся на
пов-ти тв.К,преодолевая энерг.барьер
Еадс.Выделяя избыток энергии в форме
т-ты адс-ции,м-ла может перейти в более
уст-вое адсор-ное сост.Аадс.На новом
кат-ком центре или под воздействием
актив.центра К м-ла А может перейти в
электронно-возбуждённое сост с переходом
на энергетический барьер высотою
Еист.(А*адс).Возбуждённые м-лы А,скатываясь
в ложбину продукта р-ции,превращсяв
прод.р-ции.При этом м-ла В переходит в
адсорбир.сост.Для десорбции м-лы В с
актив.ц. ей необх-мо преодолеть энергию
ЕВадс.После чего м-ла В может уйти с
каталит.центра в объём реакцион.смеси.
68.Основные теории катализа: мультиплетная, ансамблей, электронная, радикальная.
1.Мультиплетная. Основные положения этой теории:
1.Активный центр кат-ра представляет собой совокупность определенного числа адсорбционных центров, расположенных на поверхности в геометрическом соответствии со строением мол-лы, претерпевающей превращение(принцип геометрического или структурного соответствия).
2.При адсорбции реагирующих мол-л на активном центре образуется мультиплетный комплекс, в рез-те чего происходит перераспределение связей, приводящее к образованию продуктов реакции.
Для различных реакций число адсорб.центров в активном центре принимается равным 2,3,4,6.подобные акт.центры были названы дублетами,триплетами,квадруплетами,секстетами, а в общем мультиплетами.
Второе исх.положение явл. принцип энергетического соответсвия (Баландин),согласно к-ому энергия активации гетерог.р-ции явл.сложной вел-ной,в первом приближении состоящей из 2х слагаемых,одно из к-ых зависит только от энергии связи между составными частями реагир.в-в,а другое-от энергии вз-ия между катализатором и составными частями мультиплетного комплекса.Гетерог.р-ция представлена след.образом:
Исх.в-ва+кат-тор→мультипл.комплекс→кат-тор+продукты р-ции
Для I стадии необходима энергия разрыва связей,при этом выделяется энергии образования мультипл.комплекса.Е=разность этих энергий.
Для II стадии необх.энергия для разрыва связей в мультипл.комплексе. выделяется энергия обр-ния конечных продуктов.Разность этих энергий определяет скорость II стадии.
2.Ансамблей
Основное исх.положение – носителем каталит.активности явл. находящаяся на пов-ти атомная(докристаллич) фаза кат-ра,относительно к-ой пов-ть носителяч(или кристалл.фаза самого кат-ра) выполняет функцию инертной подкладки. Для каждого данного процесса акт.центром явл. ансамбль из определенного числа n атомов кат-ра.
Теория акт.ансамблей дает возможность,исходя из опытной зав-ти активности от концентрации кат-ра на пов-ти носителя,определить число n атомов в акт.центре,число Zn, областей миграции и абсолют.производительность rn акт.центра для данного процесса.
Зав-ть
активности от концентрации
3.Электронная
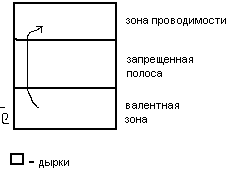 Для
полупров-ков.Эл-ны-носит.эл-ва, при
перемещ.в тв.теле остав.дырки(вакан.орбит.)
Ток возможен если недостат.эл-ов в
связях или в реш.есть изб.эл-нов. При
электрич-ве эл-ны и дырки движ.в
разл.направл. Они вызыв.превращ.акцепт.и
дон.
Для
полупров-ков.Эл-ны-носит.эл-ва, при
перемещ.в тв.теле остав.дырки(вакан.орбит.)
Ток возможен если недостат.эл-ов в
связях или в реш.есть изб.эл-нов. При
электрич-ве эл-ны и дырки движ.в
разл.направл. Они вызыв.превращ.акцепт.и
дон.
4.Радикальная
На пов-ти мет.и п/п сод.радикалоподобные состояния ат.и мол. (нескомпенсир.вал-сти) Радикал(вал-сть) явл-ся нициат. цепи(обозн.V) Реакция .вида: A2+B2= 2AB
Стадия а: A2+V AV + A (инициир.цепи)
Стадия б: A + V (A) связыв.атома вак.вал-стью
Стадия в: (A) + AV A2 + V регенерация радикала
Стадия г: B2+ V BV + (B)
Стадия д: (B) + BV B2+ V
Стадия е: (B) + V2 B +V ((В) может также реаг.на акт.ц. А):
Стадия ж: (A) + BV AB+V
Стадия з: (B) +AV AB +V; c А и В реаг.физич.адсорб.А2иВ2:
Стадия и: (A) + (B2) AB + (B)
Стадия к: (B) +(A2) AB + (A)
Обрыв цепи-путем рекомб.своб.вал-стей:
(A)+(A) = A2
(B)+(B) = B2
((A)+(B) = AB
V + V = V2 ск-сть отдельн.стадий р-ции завис.от велич.эн-гии связи A-V, B-V и теплот р-ций на стадиях а и г (Qav, Qbv). Теплоты вз-ия центров с мол-ми А2 и В2 опр-ся по разности между энергиями связей Qav и Qaa и Qbb
q1=Qav-Qaa
q`2=Qbv-Qbb
Для кажд.пары эн-гии св. Qaa и Qbb пов-сть тв.кат.должна облад.меньшими велич. Е св. атомов с кат.ц. Qav и Qbv.
69. Основные принципы твердых катализаторов.
Твердые катализаторы имеют определенный физические свойства, основными из которых являются удельная поверхность (м2/г), объем (мл/г) и структура пор, плотность (истинная и насыпная). Высокая удельная поверхность катализаторов (обычно от 10 до сотен м2/г) достигается за счет развитой системы пор, размером 1-10 нм (микропоры) и 100-1000 нм (макропоры).
По типу соединений твердые катализаторы могут быть металлами, применяются также оксиды, гидриды, сульфиды, карбиды, нитриды, а также их смеси. Кроме того, возможно применение солей и твердых кислот (особенно гетерополикислот).
Различают катализаторы массивные и нанесенные на носитель. Использование носителя основано на том, что в твердом катализаторе «работает» главным образом, тонкий слой поверхности, в котором имеются так называемые активные центры. Поэтому часто каталитически активную массу наносят на носитель, что позволяет снизить затраты на обычно более дорогой активный компонент, увеличить его дисперсность, часто также улучшаются и другие физические свойства, например, механическая прочность, теплопроводность и др.).
Обычно катализатор представляет собой смесь каталитически активного материала (например, металла) с добавкой промотора на носителе. Носитель используется для увеличения дисперсности частиц катализатора, доступности активных центров, улучшению пористой структуры. Носитель не обладает собственной каталитической активностью и не взаимодействует с каталитически активным материалом. Поэтому в качестве носителей обычно используют химически инертные, тугоплавкие оксиды с развитой поверхностью, например, активный углерод, диоксид кремния, оксид алюминия, алюмосиликаты, оксиды магния, цинка и др. Удельная поверхность носителей обычно заключается в диапазоне от 10 до 400 м2/г.
Кроме того, твердые катализаторы могут содержать промоторы. Промотор, как и носитель, не обладает каталитической активностью, но взаимодействует с каталитически активным материалом и в малых добавках улучшает некоторые эксплуатационные свойства катализатора. Например, добавка 0,2% рения (и хрома) к платине сильно повышает селективность и стабильность катализатора ароматизации в отношении спекания. Добавка 1-2% щелочи (К2О) к железным катализаторам синтеза увеличивает выход аммиака, а в синтезе Фишера-Тропша улучшают селективность по высшим углеводородам и спиртам (подавляют метанирование).
6.3.2 Основные типы твердых катализаторов
Рассмотрим теперь основные типы катализаторов, используемые в различных промышленных процессах. Для процессов кислотно-основного катализа, таких как крекинг, гидратация, дегидратация, изомеризация и др. катализатор должен обладать способностью передавать протоны реагентам. Поэтому лучшими катализаторами будут сильные кислоты, такие как цеолиты в Н-форме, алюмосиликаты, кислоты, в том числе суперкислоты и ГПК.
Для процессов гидро-дегидрогенизации, таких как гидрирование ненасыщенных соединений, гидрообессеривание нефтепродуктов, дегидрирование парафинов в олефины, ароматизации катализатор должен обладать способность к активации водорода. Обычно в качестве катализаторов таких процессов используют металлы VIII группы их оксиды и сульфиды.
В другом важном классе окислительных процессов обычно используют катализаторы – оксиды переходных металлов, например, оксиды ванадия, меди, хрома, молибдена, марганца и др.