

Билет 1.
1. Активность, коэффициент активности и способы их определения. Ограниченная и полная взаимная растворимость компонентов в различных фазовых состояниях. Диаграммы состояния.
Активность – это та концентрация, которую имел бы компонент воображаемого идеального раствора, обладающего теми же термодинамическими свойствами, что и данный реальный раствор. Она имеет размерность концентрации (моль/л).
Активность ионов – эффективная (кажущаяся) концентрация ионов с учетом электростатического взаимодействия между ними в растворе. Активность отличается от общей концентрации на некоторую величину. Отношение активности (a) к общей концентрации вещества в растворе (c, в моль/л), то есть активность ионов при концентрации 1 моль/л, называется коэффициентом активности:
γ=a/c
Коэффициент активности ионов в растворах элетролитов могут служить мерой электростатических взаимодействий в системе. Для идеальных растворов электростатические взаимодействия пренебрежимо малы, активности равны равновесным концентрациям и γ=1.
Методы расчета: так как электростатические взаимодействия особенно сильны в растворах электролитов, то основное внимание мы уделим расчетам коэффициентов активности и вычисляемой по уравнению:
![]() ,
,
где z - заряд иона; Ci - концентрация ионов одного вида в исследуемом растворе.
Таким образом, ионная сила учитывает электростатическое влияние всех ионов в растворе. Она имеет размерность концентрации и для растворов сильных 1-1 –зарядных электронов численно равна ей.
Коэффициенты активности отдельных ионов можно оценить по приближенным формулам Дебая-Хюккеля:
![]() (I≤0,01
M)
(I≤0,01
M)
![]() (I
= 0,01 -0,1 M)
(I
= 0,01 -0,1 M)
A = 0,5 при 298К; B = 0,33 при 298К., а – эмпирическая константа, учитывающая размеры ионов и характеризующая среднее расстояние сближения сольватирующих ионов в предположении, что они являются жесткими сферами.
Уравнение
Девиса при I=0,1
– 0,5 M
:
![]() ,
где а и С – константы (подбирают
эмпирически для каждого конкретного
электролита)
,
где а и С – константы (подбирают
эмпирически для каждого конкретного
электролита)
Экспериментально определить коэффициенты активности отдельных ионов невозможно, так как нельзя получить раствор, содержащий ионы только одного сорта. Можно рассчитать средий коэффициент активности:
![]()
При
высоких значениях I
z=0
:
![]() ,
С- солевой коэффициент
,
С- солевой коэффициент
Растворимость компонентов:
Полная (неограниченная) смешиваемость.
Ограниченная смешиваемость.
Практически полная нерастворимость.
Примером ограниченной смешиваемости является система фенол – вода:
Чаще всего наблюдается нижняя критическая температура так триэтиламин и вода смешиваются во всех отношениях ниже 18 0С, а выше этой температуры происходит резко выраженное расслоение смесей. Нижняя критическая точка имеется и у системы вода – 2,4,6 триметилпиридин (5,7 0С, 82,8 % масс воды). В системе никотин – вода наблюдаются обе критические точки – 60,8 0 и 208,0 0С и 34 % масс никотина.
Полной нерастворимости жидкостей друг в друге не существует, вопрос только в методах обнаружения малых примесей. Даже растворимость воды в ртути составляет величину примерно 10 -5 моль/л или около 10 –6 % по массе.
Общее давление пара в системе двух несмешивающихся жидкостей равно сумме давлений пара чистых жидкостей
![]()
следовательно такая система будет кипеть при температуре ниже. чем температура кипения самой легкокипящей. Так бензальдегид имеет температуру кипения 178 0С, а в смеси с водой перегоняется при 98 0С. Массовое отношение компонентов в дестиллате можно легко рассчитать по уравнению:

где т – массовые количества компонентов в паре, М – их мольные массы. По этому уравнению легко рассчитать необходимое количество воды, если осуществлять перегонку с водяным паром.
Нет принципиальной разницы между растворами газов в жидкостях и растворами жидкостей в жидкостях.Растворение сопровождается уменьшением объема системы и выделением теплоты порядка величины теплоты конденсации газа в жидкость.
Опытным путем был
установлен закон Генри (1803 г.): при
постоянной температуре растворимость
данного газа в данном растворителе
прямо пропорциональна его давлению над
раствором : р2
= кх2
(чем больше к
тем меньше растворимость данного газа
при данном давлении). Растворимость
газа, как видно, сильно растет с ростом
давления, и обычно уменьшается с ростом
температуры. Если газ и жидкость образуют
идеальный раствор во всей области
концентраций, то
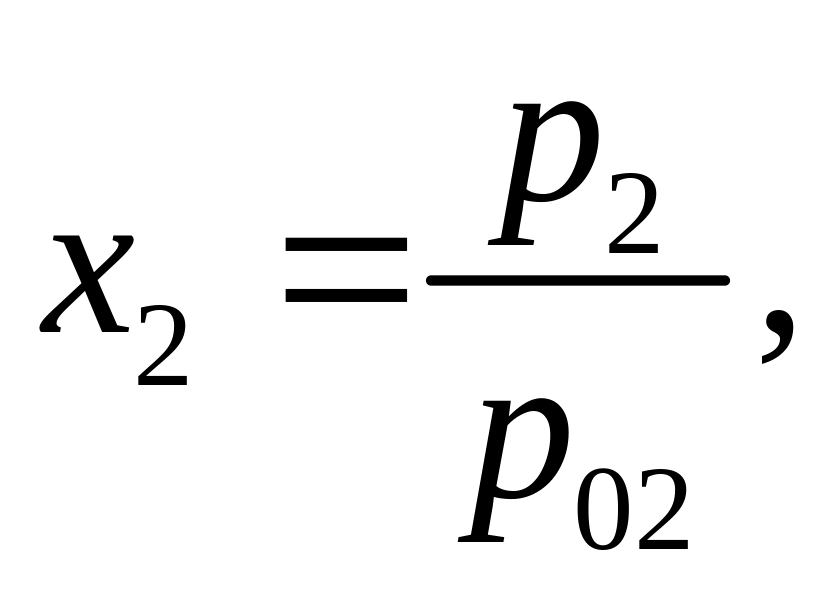 где
р02
– давление чистого газа 2 в равновесии
с его жидкой фазой при температуре
раствора, откуда видно, что мольная доля
газа в идеальном растворе не зависит
от природы растворителя. Для вычисления
р02
необходимо экстраполировать давление
насыщенного пара чистой жидкости 2 на
температуру раствора. Например,
экстраполяция давления пара жидкого
метана на температуру 298 К дает р02
= 370 атм.,
откуда получаем для
где
р02
– давление чистого газа 2 в равновесии
с его жидкой фазой при температуре
раствора, откуда видно, что мольная доля
газа в идеальном растворе не зависит
от природы растворителя. Для вычисления
р02
необходимо экстраполировать давление
насыщенного пара чистой жидкости 2 на
температуру раствора. Например,
экстраполяция давления пара жидкого
метана на температуру 298 К дает р02
= 370 атм.,
откуда получаем для
![]() идеальную растворимость х2
= 0,0027. Для сравнения растворимость СН4
в гексане
равна 0,0031, в ксилоле 0,0026.
идеальную растворимость х2
= 0,0027. Для сравнения растворимость СН4
в гексане
равна 0,0031, в ксилоле 0,0026.
Для реальных растворов сопоставление растворимости различных газов в одинаковых условиях не позволяет установить простой закономерности. В общем здесь действует правило: подобное растворяется в подобном, т.е. полярные газы легче растворяются в полярных растворителях, чем неполярных. Далее неполярные газы, обладающие более высокой критической температурой, т.е. легче сжижаемые, легче растворяются в неполярных жидкостях, чем газы сжижаемые труднее.Если происходит химическое взаимодействие газа и растворителя, как, например, в растворах H2S, CO2, HCl, NH3 в воде , то растворимость резко повышается.
Если в одном и том же растворителе одновременно растворяются два газа, невзаимодействующие химически друг с другом и с растворителем, то они не оказывают влияние на растворимость друг друга. Заметим, что кислород примерно в два раза более растворим в воде, чем азот, что оказывается важным для организмов, живущих в воде. При цикле последовательных растворений и выделений воздуха, выделяющийся газ постепенно обогащается кислородом
Растворимость газов в воде обычно уменьшается при добавлении других растворимых веществ, в частности электролитов. Это явление, получившее название «высаливания», впервые было обнаружено И.М.Сеченовым. Он получил уравнение, выражающее влияние концентрации электролита на растворимость газов
ln C =ln C0 – bm,
где С0 – растворимость данного газа в чистой воде, С – растворимость его при той же температуре в растворе электролита, имеющим концентрацию « т » (моль/л),
в – эмпирическая постоянная, зависящая от природы газа, электролита и температуры. Ионы электролита как бы уменьшают из-за гидратации массу «свободной» воды.
Диаграмма состояния (фазовая диаграмма), графическое изображение всех возможных состояний термодинамической системы в пространстве основных параметров состояния температуры Т, давления р и состава х (обычно выражаемого молярными или массовыми долями компонентов). Для сложных систем, состоящих из многих фаз и компонентов, построение диаграмм состояния является единственным методом, позволяющим на практике установить, сколько фаз и какие конкретно фазы образуют систему при данных значениях параметров состояния. Каждое реально существующее состояние системы на диаграмме состояния изображается так называемой фигуративной точкой; областям существования одной фазы отвечают участки пространства (на трехмерных диаграммах состояния) или плоскости (на двухмерных диаграммах состояния), условиям сосуществования фаз - соотв. поверхности или линии; изменение фазового состояния системы рассматривается как движение фигуративной точки на диаграммах состояния. Анализ относительного расположения объемных участков, поверхностей, линий и точек, которые образуют диаграммы состояния, позволяет однозначно и наглядно определять условия фазового равновесия, появления в системе новых фаз и химических соединений, образования и распада жидких и твердых растворов и т. п. Диаграммы состояния. используют в материаловедении, металлургии, нефтепереработке, хим. технологии (в частности, при разработке методов разделения веществ), производствах электронной техники и микроэлектроники и т. п. С ее помощью определяют направленность процессов, связанных с фазовыми переходами, осуществляют выбор режимов термообработки, отыскивают оптимальные составы сплавов и т. п. Теоретическими основами построения и интерпретации диаграмм состояния равновесных систем являются: 1) условие фазового равновесия, согласно к-рому хим. потенциалы mi каждого i-го компонента во всех фазах при равновесии равны; 2) условие химического равновесия, согласно к-рому сумма хим. потенциалов вступающих в р-цию в-в при равновесии равна аналогичной сумме для продуктов р-ции; 3) фаз правило Гиббса, согласно к-рому число компонентов К, число фаз Ф и вариантность системы v (т. е. число независимых параметров состояния, к-рые можно в определенных пределах изменять без изменения числа и природы фаз) связаны соотношением: v = К — Ф + 2. Цифра 2 означает, что учитываются только два интенсивных параметра состояния - т-ра и давление. Если учитываются и др. параметры, напр., напряженности электромагнитного или гравитационного полей, вариантность системы соотв. увеличивается. Различают нонвариантные (v = 0), моновариантные (v = 1), дивариантные (v = 2) и т. д. состояния (равновесия); 4) правило о соприкасающихся пространствах состояния, согласно к-рому если два разных пространства состояния (поля в случае плоской диаграммы) соприкасаются по линии, то они различаются между собой на одну фазу, если поля соприкасаются в точке, то состояния различаются на две фазы. Для построения Д. с. расчетным путем необходимо знать зависимости хим. потенциалов всех компонентов системы от Т, р и состава фаз. Приближенные методы расчета с применением ЭВМ интенсивно развиваются, в частности, для многокомпонентных сплавов. Однако пока диаграммы состояния строят на основе экспериментальных данных, получаемых главным образом термическим анализом, которыйрый позволяет определять зависимости температур плавления или кристаллизации от состава, а также изучением равновесий жидкость - пар и жидкость - жидкость. Широко используют рентгеновский фазовый анализ, данные о микроструктуре затвердевших расплавов, измерения физ. свойств фаз (см. Диаграмма состав-свойство). Изучение диаграмм состояния составляет основное содержание физико-химического анализа.
Однокомпонентные системы. Однокомпонентной системой является любое простое вещество или химическое соединение, обладающее строго определенным составом в газообразном, жидком и твердом состояниях. Диаграммы состояния обычно строят на плоскости в координатах Т-р (рис. 1). Фазовые поля (области существования) пара V, жидкости L и твердой фазы S дивариантны, т.е. допускают одновременное изменение двух параметров состояния - Т и р.
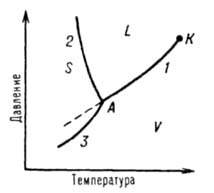
Рис. 1 Диаграмма состояния однокомпонентной системы. S, L и V - соотв. области существования твердой, жидкой и паровой фаз; 1, 2 и 3 кривые кипения (испарения), плавления и возгонки (сублимации) соотв., К критич. точка; А тройная точка.
Двухфазное равновесие между жидкостью и паром изображается кривой кипения (или испарения) 1, между жидкостью и кристаллами - кривой плавления 2, между кристаллами и паром - кривой возгонки (или сублимации) 3. Все двухфазные равновесия моновариантны, т. е. не нарушаются при произвольном изменении только одного из параметров, Т или р; при этом значение другого определяется из Д. с. Кривая кипения 1 характеризует зависимость давления насыщенного пара вещества от температуры или зависимость температуры кипения вещества от внешнего давления. Со стороны повышенных T и р эта кривая заканчивается в критической точке К, где исчезает различие в свойствах между жидкостью и ее паром (подробнее см. ст. Критическое состояние). Жидкость может находиться в переохлажденном состоянии (пунктирная линия на рис. 1). Аналогично кривая плавления характеризует зависимость т-ры плавления от внешнего давления, кривая возгонки - температурную зависимость давления насыщенного пара над твердым веществом. На рис. 1 ход кривой плавления соответствует повышению т-ры плавления с ростом давления, однако возможно и понижение температуры плавления с давлением (см. Клапейрона - Клаузиуса уравнение). Все три кривые моновариантных фазовых равновесий сходятся в тройной точке А, отвечающей параметрам состояния, при которых находятся в нонвариантном равновесии три фазы. Диаграммы состояния усложняется, если вещество в твердом состоянии может существовать в различных кристаллических модификациях. Каждой модификации отвечает свое фазовое поле. Линии моновариантных равновесий, разграничивающие эти поля, наз. кривыми превращений.
Двойные системы. Состояние двойной системы определяется тремя независимыми параметрами - Т, р и содержанием х одного из компонентов, поэтому Д. с. такой системы трехмерна. Обычно принимают постоянными Т или р и рассматривают соответствующие плоские сечения Д. с., называемые соотв. изотермич. (р — х)или изобарными (Т — х)Д. с. В конденсир. системах роль давления сравнительно невелика и в качестве параметров состояния обычно выбирают Т и состав (концентрацию одного из компонентов).
Диаграммы плавкости. Такие диаграммы состояния служат для установления условий равновесия между твердыми и жидкими фазами. Рассмотрим их основные типы. Простейший вариант соответствует случаю, когда компоненты А и В образуют одну жидкую фазу (расплав или раствор), при охлаждении которой только выделяются (кристаллизуются) индивидуальные вещества (не образуются ни твердые растворы, ни хим. соединения).
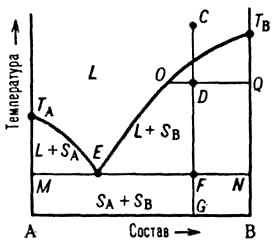
Рис. 2. Диаграмма плавкости двойной системы, компоненты к-рой А и В не образуют твердых р-ров. L - область существования жидкости (расплава), (L + SA) и (L + SB) области сосуществования жидкой фазы и твердых А и В соотв.; (SA + SB) область существования мех. смеси твердых А и В. ТАETВ и MEN - линии ликвидуса и солидуса соотв., E-эвтектич. точка. С, D, F, G, О и Q фигуративные точки (пояснения в тексте).
Тройные системы. Состояния тройных систем однозначно определяются четырьмя независимыми параметрами: Т, р и молярными (массовыми) долями двух компонентов (доля третьего компонента определяется из условия равенства единице суммы долей всех компонентов). Поэтому при построении Д. с. тройных систем один из независимых параметров (р или Т) или два (р и T) фиксируют и рассматривают пространственные изобарные или изотермич. диаграммы или плоские изобарно-изотермич. диаграммы, соответствующие одному из сечений пространственной Д. с. Каждому составу тройной смеси отвечает определенная точка на плоскости составов. Область возможных составов тройных систем наз. композиционным треугольником или треугольником составов. В системе прямоугольных координат он представляет собой прямоугольный равнобедренный треугольник, вершины к-рого отвечают компонентам А, В и С, а стороны - двойным смесям АВ, ВС и СА. Более распространено использование равностороннего композиц. треугольника. В этом случае все компоненты равноправны, а за начало координат можно с равным основанием принять любую из его вершин (см.