

48.Кондуктометрия,потенциометрия.
Кондуктометрия - это совокупность электрохимических методов анализа, основанных на измерении удельной электропроводности (или сопротивления) растворов электролитов.
Любое вещество характеризуется своим электрическим сопротивлением (R). Величина обратная сопротивлению называется электропроводностью или электрической проводимостью (G). Для раствора электролита, находящегося между двумя электродами, площадь поверхности которых равна S и расстояние между которыми равно :



где - удельная электропроводность раствора/
Удельная электропроводность связана с молярной концентрацией эквивалента вещества (моль/л):
= 110-3С,
Молярная
электропроводность равна произведению
абсолютной скорости движения иона на
постоянную Фарадея. При уменьшении
концентрации электролита и уменьшении
ионной силы скорости движения ионов
возрастают, поэтому величина
увеличивается. При бесконечном разбавлении
молярная электропроводность достигает
некоторого предельного (ненулевого)
значения, называемого предельной
молярной электропроводностью .
Согласно закону
Кольрауша:
.
Согласно закону
Кольрауша:


Контактные кондуктометрические измерения проводят в ячейке для измерения электропроводности. Простейшая ячейка представляет собой стеклянный сосуд с двумя плоско-параллельными платиновыми электродами. Для уменьшения концентрационной поляризации используют платинированную (покрытую платиновой чернью) платину, имеющую большую площадь поверхности. Раствор, находящийся в ячейке, постоянно перемешивается. Ячейку подключают к источнику переменного тока, имеющего частоту около 1000 Гц. Непосредственно измеряемой величиной в кондуктометрии является не электропроводность, а сопротивление. Сопротивление раствора можно измерять с помощью моста Уитстона Мосты переменного тока могут быть уравновешенными и неуравновешенными. В случае уравновешенного моста величины сопротивлений R1, R2 и R3 должны быть такими, чтобы мост пришёл в состояние равновесия, при котором сила тока в измерительной диагонали равна нулю (илиимеетминимальноезначение).

Прямая кондуктометрия основана на существовании (в области разбавленных и умеренно концентрированных растворов) прямолинейной зависимости между и С. Поскольку электропроводность раствора является аддитивной величиной, прямая кондуктометрия обладает малой избирательностью и используется лишь в тех случаях, когда достаточно знать общую концентрацию ионов в растворе, при контроле качества воды, определении суммарного содержания солей в природных водах или биологических жидкостях. Кондуктометрический детектор является одним из детекторов, используемых в ВЭЖХ. Прямую кондуктометрию используют также для определения различных физико-химических характеристик вещества (Ka, KS и др.).
Кондуктометрическое титрование основано на изменении удельной электропроводности раствора в зависимости от количества добавленного титранта. Чаще всего в кондуктометрическом титровании используются протолитические реакции, реже всего - окислительно-восстановительные. Электропроводность исходного раствора должна заметно отличаться от электропроводности реагента или продукта реакции. Константу ячейки при кондуктометрическом титровании знать не обязательно, поскольку определяют не абсолютное значение , а её изменение в процессе титрования.
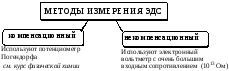
Потенциометрическими называют методы анализа, основанные на измерении зависимости равновесного электродного потенциала от активности определяемого иона.При потенциометрических измерениях используется электрохимическая ячейка, работающая в режиме гальванического элемента. В состав ячейки входит индикаторный электрод, потенциал которого зависит от активности определяемого иона или от активности хотя бы одного из компонентов протекающей химической реакции, и электрод сравнения (чаще всего хлоридсеребряный), величина потенциала которого постоянна. Величина потенциала индикаторного электрода связана с активностью определяемого иона уравнением Нернста (см. главу 7).



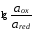 (при
298 К)
(при
298 К)


Измерение ЭДС гальванического элемента проводят в условиях, которые близки к термодинамическим:
• сила тока, протекающего через ячейку, должна быть равна 0;
• время, в течение которого проводится измерение, должно быть достаточным для достижения равновесия.

Прямая потенциометрия
В прямой потенциометрии концентрацию (активность) опреде-ляемого вещества рассчитывают, исходя из величины ЭДС гальванического элемента. Чаще всего индикаторным в прямой потенциометрии является ионоселективный электрод. Прямые потенциометрические измерения, в которых используется ионоселективный электрод, называются ионометрией. Данный метод анализа характеризуется простотой и экспрессностью методик, недорогой аппаратурой.
Потенциометрическим титрованием называется метод анализа, основанный на регистрации изменения потенциала индикаторного электрода в процессе химической реакции между определяемым веществом и титрантом.
В основе потенциометрического титрования могут лежать различные протолитические, окислительно-восстановительные, осадительные реакции и реакции комплексообразования, протекающие количественно, стехиометрично и с приемлемой скоростью. Выбор индикаторного электрода для выполнения потенциометрического титрования зависит от используемой реакции. Конечную точку титрования обнаруживают с использованием кривой титрования, её производных, а также методом Грана.
Хроматографические методы индинтификации лекарственных веществ
Для идентификации в-в (качественного определения ) используются: газовая хроматография, жидкостная хроматография(различные её виды)
В хроматографии для идентификации веществ используются следующие характеристики (характеристики удерживания) :
Время от момента ввода пробы до момента регистрации максимума пика называется временем удерживания (tR). tR = tm + ts
Время удерживания не зависит от концентрации вещества, но зависит от его природы, а также от природы подвижной и неподвижной фазы и условий хроматографирования. Время удерживания вещества зависит от упаковки сорбента и поэтому может изменяться при переходе от одной колонки к другой.
Более надёжной характеристикой является исправленное время удерживания ( tR’),
которое равно разности между временем удерживания данного вещества и несорбируемого компонента (t0). Поскольку t0 = tm, то tR’ = tS.
Объём подвижной фазы, который необходимо пропустить через колонку с определённой скоростью для того, чтобы элюировать вещество, называется удерживаемым объёмом (VR). Аналогично понятию исправленное время удерживания существует понятие исправленныйудерживаемый объём (VR’ ) . VR= tR× F VR’=( tR- tm)× F = VR- Vm
где F - объёмная скорость подвижной фазы (см3/мин)
Отношение
равновесной концентрации вещества в
неподвижной фазе (Cs)
к его равновесной концентрации в
подвижной фазе (Сm)
называется коэффициентом распределения
(D).
![]()
Удерживаемый
объём связан с коэффициентом
распределения уравнением, называемым
основным уравнением хроматографии:
![]()
Произведение коэффициента распределения на соотношение объёмов неподвижной и подвижной фазы называется коэффициентом ёмкости колонки ( k’ ):
![]()
![]()
Так же используется параметр, называемый индексом удерживания. В газовой хроматографии для определения индекса удерживания в качестве стандартов берут два соседних н-алкана, один из которых элюируется до исследуемого соединения, а второй после. Логарифмический индекс удерживания равен:
 где
z - число атомов углерода в молекуле
н-алкана, который элюируется первым .
где
z - число атомов углерода в молекуле
н-алкана, который элюируется первым .
Затем по справочным таблицам можно определить круг веществ, которые имеют близкую к рассчитанной величину индекса Ковача.
При Анализе плоскостной хроматограммы используются свои характиристики.
Разделяемые компоненты образуют на хроматографической пластинке или полоске хроматографической бумаги отдельные зоны (пятна).

Положение
отдельных хроматографических зон на
хроматограмме характеризуют с помощью
величины Rf,
равной отношению расстояния, пройденного
зоной вещества от стартовой линии до
центра зоны (x), к расстоянию от
стартовой линии до границы фронта
растворителя к концу опыта (L)![]()
Величина Rf может принимать значения от 0 до 1. Если Rf = 0, то вещество остаётся на старте, если Rf = 1, то оно поднимается с фронтом растворителя. Величина Rf является качественной хроматографической характеристикой вещества. Она зависит от природы вещества, подвижной и неподвижной фазы, условий хроматографирования и, в определённых пределах, не зависит от концентрации вещества. Подвижность разделяемых веществ можно также сравнить с подвижностью вещества, принятого за стандарт
![]()
Величина Rf связана с коэффициентом распределения вещества (D) и коэффициентом емкости неподвижной фазы по отношению к данному веществу (k’ ) следующими уравнениями:
![]()
![]()