

Акушерство для студентов / Запоражан том 1
.pdfРозділ 6. Пренатальний скринінг...
номегалію, водянку і смерть плода. Електрофорез гемоглобіну виявляє відсутність HbF, HbA і 90–100 % гемоглобіну Hbα 4, який відомий ще під назвою гемоглобіну Барта. За наявності гемоглобіну Н (HbH хвороба) має місце делеція гена 3α глобі ну, що призводить до надмірного накопичення β ланцюгів у еритроцитах. Бета тет рамери є нестабільними і швидко підлягають окисленню, що призводить до дест рукції клітинних мембран. Такі діти народжуються з анемією; при електрофорезі гемоглобіну виявляють гемоглобін Барта і деяку кількість HbH. Через декілька місяців гемоглобін Барта зникає і виявляють гемоглобін А і гемоглобін Н. При наяв ності двох делецій фенотипові прояви α таласемії є менш вираженими і проявля ються мікроцитарною анемією. Пацієнти з 1 мутацією є безсимптомними носіями, діагноз підтверджується за допомогою генетичного аналізу (генетичне картування).
Скринінг на α таласемію, як і на β таласемію проводиться лише у групах ризику: за наявності мікроцитарної анемії виконують електрофорез гемоглобіну. Наявність cis або trans мутацій є дуже важливою. Якщо обидва партнери мають cis мутацію, їх дитина матиме 25 % ризику тяжкого захворювання, що може призвести до смерті. Якщо обидва батьки є носіями trans мутації, дитина також матиме trans мутацію (безсимптомний носій).
Діагностика моногенних дефектів інколи може бути можливою при ультразвуко вому дослідженні (наприклад, ехогенні кишки при кістозному фіброзі) або при ви конанні специфічних молекулярно діагностичних тестів (FGFR2 рецептори факто ра росту 2 фібробластів при синдромі Аперта (краніосиностозі); FGFR3 — при тана тофорній дисплазії і ахондроплазії).
Молекулярні методи діагностики генетичних захворювань включають такі мето
ди:
1.Прямий аналіз мутацій є найбільш точним методом. Полімеразна ланцюгова реакція (ПЛР) в агарозному гелі використовується для ампліфікації ділянки ДНК, що містить мутацію (делецію, інверсію або дуплікацію). Цей метод часто викорис товують для діагностики м’язової атрофії Дюшенна.
2.Алель?специфічний однонуклеотидний аналіз, що часто використовується для ідентифікації точкових мутацій. ПЛР продукт розміщується на фільтрі та гібриди зується з однонуклеотидними зразками нормального препарату і препарату з мута цією. У випадку мутації він зв’язується з мутантним однонуклеотидним зразком і дає позитивний сигнал.
3.Аналіз поліморфізму довжини рестриктивного (обмежувального) фрагмента ви користовується, якщо мутація утворює або руйнує місця розпізнавання дефіцитних ензимів. ПЛР продукт інкубується з дефіцитним ензимом і аналізується в агарозно му гелі. За наявності мутації ДНК фрагмент, утворений при вивільненні специфіч ного ензима, є аномальним за розміром і мобільністю гелю.
4.Зчеплений аналіз використовується при відомій мутації гена, але якщо ген і мутація не характеризовані. ДНК маркер підводиться до гена для виявлення мож ливості трансмісії ураженого гена і ризику його успадкування.
Мультифакторіальні спадкові захворювання
Мультифакторіальні спадкові захорювання збільшуються серед родичів і реци дивують не за менделівськими принципами передачі генів. Вади розвитку часто сто суються одного органа або збільшуються у представників однієї статі. Типовими прикладами мультифакторіального успадкування є дефекти нервової трубки, щіли на губи і піднебіння, природжені вади серця. Ризик рецидивів мультифакторіальних
189
Акушерство і гінекологія. Том 1
захворювань за наявності захворювання в 1 родича першого ступеня спорідненості становить 2–3 %, при захворюванні у 2 родичів — збільшується до 4–6 %. Неура жені носії мають 1–2 % ризику у хворих нащадків. Аномалії, які мають мультифак торіальні риси, можуть бути компонентами генетичних синдромів з різним прогно зом і різною частотою рецидивів. Так. наприклад, дефекти нервової трубки можуть бути асоційовані з полікістозом нирок і полідактилією, що в сукупності становить синдром Мекеля — аутосомно рецесивне захворювання з частотою рецидивів 25 %.
Природжені вади розвитку плода
Природжені вади розвитку можуть зустрічатись у будь якій системі та органі плода, що залежить від часу дії тератогенного фактора на плід. Тератогени можуть бути представлені медикаментозними препаратами, інфекційними агентами, особ ливо вірусними, які можуть проходити через плаценту і викликати внутрішньоут робну інфекцію у плода, хімічними чи фізичними чинниками (ртуть, іонізуюче ви промінювання тощо).
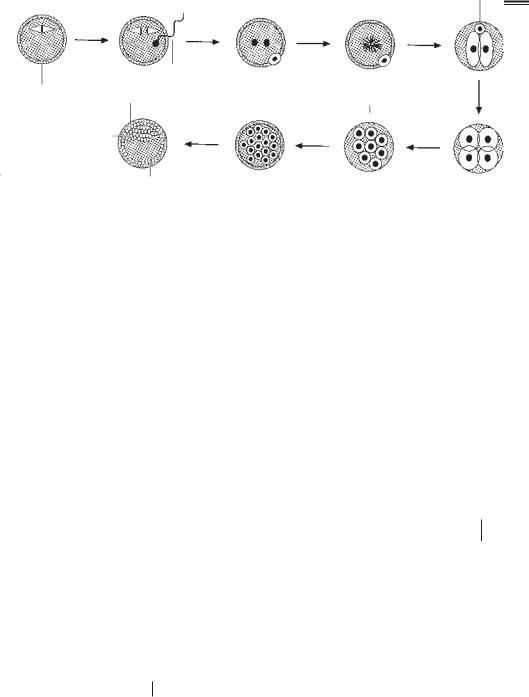
Після фертилізації яйцеклітини сперматозоїдом, утворена зигота підлягає декіль ком клітинним діленням і досягає стадії 16 клітинної морули на 4 й день розвитку (рис. 6.1). Після досягнення морулою порожнини матки, в ній відбувається вилив рідини, що розділяє морулу на внутрішній і зовнішній шари — внутрішню і зовніш ню клітинну масу, і утворюється бластоциста. Бластоциста, в свою чергу, розді ляється на ембріобласт і трофобласт і імплантується в ендометрій наприкінці 1 го тижня розвитку. На початку 2 го тижня трофобласт починає диференціюватись у 2 шари: внутрішній, цитотрофобласт і зовнішній, синцитіотрофобласт, які разом утво рюють плаценту. Незабаром внутрішня клітинна маса утворює двошаровий зарод ковий диск, що складається з епібласта і гіпобласта.
Протягом 3 го тижня ембріонального розвитку в ембріоні відбувається процес гаструляції. Він характеризується появою первинної смужки в епібласті з подаль шою інвагінацією епібласта і формуванням трьох зародкових шарів ембріона: внут рішнього — ендодерми, середнього — мезодерми і зовнішнього — ектодерми. З ендо дермального шару розвиваються дихальна система і шлунково кишковий тракт; ме зодерма дає початок серцево судинній, скелетно м’язовій і урогенітальній системам.
3
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
1 |
7 |
4 |
||||
|
|
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5
Рис. 6.1. Ранній ембріогенез:
1 — яйцеклітина; 2 — сперматозоїд; 3 — двоклітинна стадія; 4 — морула; 5 — зовнішній клітинний шар; 6 — внутрішня клітинна маса; 7 — бластоциста
190

Розділ 6. Пренатальний скринінг...
Ектодерма дає початок нервовій системі, шкірі, волоссю, сенсорним органам (орган зору, орган слуху, орган нюху).
Найбільш уразливим періодом внутрішньоутробного розвитку щодо дії терато генів є період органогенезу, звичайно з 3 го по 8 й тиждень після запліднення (5– 10 тиж гестаційного віку), коли утворюються більшість органів і систем організму.
Дефекти нервової трубки
Утворення нервової трубки відбувається на 22–23 й день після запліднення (4 й тиждень розвитку) в ділянці 4–6 сомітів. Злиття нервової трубки відбувається в краніальному і каудальному напрямках. Передній нейропор (майбутній мозок) зак ривається на 25 й день, а задній нейропор — майбутній спинний мозок — закри вається на 27 й день. Закриття нервової трубки збігається зі встановленням її влас ного кровопостачання. Більшість дефектів нервової трубки розвивається внаслідок дефекту її закриття на 4 му тижні розвитку (6 й тиждень гестаційного віку).
Дефекти нервової трубки, зокрема аненцефалія і щілина хребта (spina bifida) є
класичним прикладом мультифакторіального успадкування, що демонструють по єднання генетичних факторів оточуючого середовища. Існують значні географічні та етичні варіації частоти цих вад розвитку.
Доведено, що зменшення рівня фолієвої кислоти в крові матері асоційовано з розвитком дефектів нервової трубки у плода. Отже, преконцепційна профілактика шляхом вживання фолієвої кислоти перед заплідненням і протягом вагітності доз воляє значно знизити частоту дефектів нервової трубки, в тому числі їх рецидивів. Крім того, ризик дефектів нервової трубки збільшується вдвічі у випадку гомозигот ності за мутацією C677T у гені, що кодує фермент метилтетрагідрофолатредуктазу зі зменшеною активністю. Але частка цих мутацій у розвитку дефектів нервової трубки не є значною. Ризик дефектів нервової трубки при певному генотипі може дуже варіювати залежно від материнських факторів ризику, зокрема рівня вітаміну В12 або фолатів у сироватці крові.
Аненцефалія (екзенцефалія) виникає внаслідок порушень процесу закриття крані альної частини нервової трубки з частотою 1:1500 новонароджених, частіше ушко джується плід жіночої статі. При цьому склепіння черепа взагалі не утворюється, а аномальний мозок залишається відкритим, дегенерує і вимивається амніотичною рідиною. Через відсутність у плода механізму ковтання ця вада розвитку супрово джується багатоводдям в останні місяці вагітності.
Плоди зі щілиною хребта (spina bifida) можуть бути виявлені при ультразвуково му обстеженні, при якому може візуалізуватися не сам відкритий спинномозковий канал, а асоційовані з ним ознаки. Так, у разі spina bifida при ультразвуковому дослі дженні виявляються ознаки «лимона» (западина лобних кісток) або «банана» (витя гання мозочка каудально і деяке його сплощення). Часто асоційованими аномалія ми можуть бути вентрикуломегалія (розширення шлуночків мозку) та зігнуті ступні. До широкого впровадження ультрасонографії у реальному масштабі часу, скринін говим тестом щодо вад розвитку нервової трубки був лише рівень α фетопротеїну (АФП) у сироватці крові матері. Відкрита нервова трубка плода призводить до збільшення рівня АФП в амніотичній рідині і, відповідно, в крові матері.
Функції новонароджених зі щілиною хребта залежать від рівня ураження спин ного мозку. Якщо щілина хребта локалізується низько у крижовій ділянці, функція сечового міхура і кишок може бути не порушеною. Але в більшості випадків наявна повна втрата функцій сечового міхура, кишок і нижніх кінцівок. Сучасні дослі дження спрямовані на можливіcть відновлення тяжких дефектів у плода in utero.
191

Акушерство і гінекологія. Том 1
Невеликі дефекти черепа, крізь які випинають тканини головного мозку (грижа головного мозку, енцефалоцелє) і/чи мозкові оболонки (грижа мозкових оболонок, менінгоцелє), є досить частими і можуть бути усунені хірургічним шляхом.
Гідроцефалія (водянка головного мозку) характеризується надмірним накопичен ням спинномозкової рідини в системі шлуночків мозку. Виникає внаслідок стенозу сільвієвого водопроводу, коли спинномозкова рідина з бічних і третього шлуночків не має відтоку в четвертий шлуночок і далі у підпавутинний простір.
Синдром Арнольда — Кіарі зумовлюється зміщенням мозочка у каудальному на прямку і його защемленням у великому отворі (foramen magnum). Ця вада супрово джує майже всі випадки кістозної щілини хребта і часто поєднується з гідроцефалією.
Мікроцефалія проявляється зменшенням склепіння черепа внаслідок аномально го розвитку мозку. Вада може передаватися за аутосомно рецесивним типом або бути пов’язаною з пренатальною інфекцією, дією медикаментозних препаратів або інших тератогенів.
Природжені вади серця
Існують численні варіанти структурних аномалій серця, що супроводжуються змінами фізіологічних основ діяльності серцево судинної системи. Розвиток серця починається на 3 му тижні після запліднення, коли ангіогенні клітинні кластери утворюються у передньоцентральній зоні ембріона. Одночасно зі згинанням ембріо на в цефалокаудальному напрямку кардіогенна ділянка також згинається і утворює серцеву трубку. Навіть у цій ранній стадії ембріональне серце вже отримує венозну кров із каудального кінця і виштовхує кров через першу аортальну дугу в дорзальну аорту. В цей час мезодерма оточує серцеву трубку трьома шарами клітин, які утво рюють рівні: зовнішній — епікард, середній — міокард і внутрішній — ендокард. Між 23 і 28 м днями серцева трубка подовжується і утворює серцеву петлю із за гальним передсердям і вузьким атріовентрикулярним з’єднанням, що сполучає його з первинним шлуночком. Цибулина серця (bulbus cordis) у каудальній частині серце вої трубки утворює три структури: проксимальна третина дає початок трабекулярній частині правого шлуночка; середня частина (conis cordis) формує вихідний тракт шлуночків, а дистальний сегмент (truncus arteriosus) зрештою утворює проксимальну частину аорти і легеневу артерію (рис. 6.2).
Між 27 і 37 м днями серце продовжує розвиватися шляхом утворення основної перегородки. Формування перегородки відбувається за рахунок тканини ендокарда (ендокардіальних валиків), яка розділяє його простір на дві порожнини. Праве і ліве передсердя утворюються завдяки розвитку первинної та вторинної перегородки, що розділяє первинне передсердя, хоча залишає міжпередсердний отвір, або овальний отвір (foramen ovale), для можливості шунта (скиду) крові справа — наліво. На 4 му тижні ендокардіальні валики знов з’являються у передсердно шлуночковому каналі й утворюють правий і лівий канали, а також мітральний і трикуспідальний клапани. Протягом цього часу медіальні стінки шлуночків поступово зливаються одна з од ною і утворюють м’язову частину міжшлуночкової перегородки. Артеріальний ко нус (conis cordis) вступає у середню частину цибулини серця, і протягом 5 го тижня розвитку ендокардіальні валики підрозділяють його на вихідні шляхи для правого і лівого шлуночків, а також на оболонкову частину міжшлуночкової перегородки. Ендокардіальні валики з’являються також в артеріальному стовбурі (truncus arteriosus)
— дистальній третині цибулини серця — і ростуть у вигляді спіралі, формуючи аор топульмонарну перегородку, розділяючи артеріальний стовбур на аортальний і пуль мональний тракт.
192
Розділ 6. Пренатальний скринінг...
5
6
4
7
9
8
3
2 |
1 |
|
1
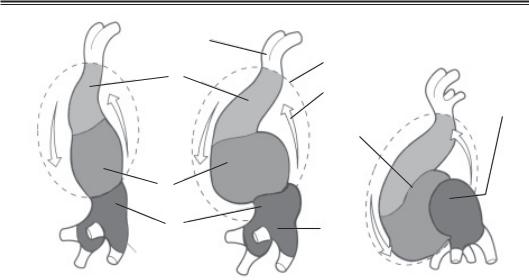
Рис. 6.2. Розвиток чотирикамерного серця плода:
1 — венозний синус; 2 — передсердя; 3 — шлуночок; 4 — цибулина серця; 5 — корінь аорти; 6 — перикард; 7 — порожнина перикарда; 8 — бульбовентрикулярна борозна; 9 — ліве передсердя
Будь який етап розвитку серця може бути порушений, що призведе до утворен ня аномалій. Так, наприклад, якщо стінки шлуночків не з’єднаються, утвориться дефект міжшлуночкової перегородки, що, за відсутності хірургічного лікування, приз веде до розвитку синдрому Ейзенменгера (гіпертрофія правого шлуночка і легенева гіпертензія, шунт крові «справа наліво»). Однією з найбільш частих вад розвитку серця є тетрада Фалло, яка включає дефект міжшлуночкової перегородки, декстра позицію аорти, стеноз або атрезію легеневої артерії та гіпертрофію правого шлуноч ка (рис. 6.3). Може зустрічатися транспозиція судин, коли аорта і легенева артерія з’єднуються в одному шлуночку. Інші аномалії включають коарктацію аорти, відкри? ту артеріальну протоку (також призводить до розвитку синдрому Ейзенменгера).
Ультразвуковий скринінг аномалій розвитку серця залежить від якості ультра звукової візуалізації серця. Багато вад розвитку, включаючи коарктацію аорти, де фект міжшлуночкової та міжпередсердної перегородки, часто неможливо виявити при ультразвуковому обстеженні.
Наслідки природжених вад серця дуже варіюють. Більшість із них підлягають хірургічній корекції. Такі вади, як гіпоплазія лівих відділів серця, звичайно є неку рабельними і хворі часто вмирають у молодому віці.
Синдром Поттера
Синдром Поттера виникає внаслідок ниркової недостатності, яка призводить до вираженого маловоддя (ангідрамніону), що, в свою чергу, спричинює гіпоплазію легенів і контрактури у плода. Під хворобою Поттера розуміють двобічну агенезію нирок. Ниркова недостатність може розвиватись у плода також внаслідок обструкції сечових шляхів.
193

Акушерство і гінекологія. Том 1
Права сонна артерія |
Ліва сонна артерія |
|
|
|
|
Права підключична |
Ліва підключична |
|
артерія |
артерія |
|
Плечо головний |
|
|
стовбур |
|
|
Верхня порожниста |
Аорта |
|
|
||
вена |
|
|
Права легенева |
|
Ліва легенева артерія |
|
|
|
артерія |
|
|
|
|
Ліві легеневі вени |
Праві легеневі |
Стеноз легеневого |
|
вени |
стовбура |
|
|
|
Дефект |
|
|
міжшлуночкової |
|
|
перегородки |
Правий шлуночок |
|
|
|
|
Лівий шлуночок |
Нижня порожниста
вена
Гіпертрофія правого шлуночка
Рис. 6.3. Тетрада Фалло
Утворення нирок відбувається з проміжної мезодерми послідовно з участю трьох ниркових систем, які поступово регресують (переднирка — pronephros, первинна нир? ка — mesonephros і остаточна нирка — metanephros), і починається на 4 му тижні розвитку. Першою є переднирка, яка утворюється у шийному відділі зародка і не функціонує. На 5 му тижні виникає первинна нирка, яка утворює мезонефральні, або є мюллерові протоки. Відросток мюллерових проток — зачаток сечовода — роз ширюється і розділяється з утворенням сечовивідної системи — трубочок, чашечок, ниркових мисок і сечоводів у чоловіків і жінок. За наявності тестостерону, мезо нефральні протоки у чоловіків також дають початок сім’явивідним протокам, при даткам яєчка, еякуляторним протокам і сім’яним пухирцям. У жінок ці додаткові протоки дегенерують, за винятком залишкової (рудиментарної) Гартнерової прото ки, що може утворювати доброякісні кісти за ходом широкої зв’язки матки. Третя ниркова система — остаточна нирка — також виникає на 5 му тижні гестації і почи нає функціонувати близько 9 го тижня. Зачаток сечовода з мезонефральних проток контактує з остаточною ниркою — метанефросом — і спонукає його до утворення нефронів. Якщо цей контакт не відбувається, має місце агенезія нирок. Прилегла дорзальна аорта віддає колатералі у метанефрос для завершення розвитку ниркових клубочків.
194

Розділ 6. Пренатальний скринінг...
До 7 го тижня клоака (проксимальна частина алантоїса, що у дистальному кінці контактує з жовтковим мішком) розділяється на 2 частини: урогенітальний синус і аноректальний канал. Протягом цього процесу каудальна (найбільша) частина мезо нефральних проток абсорбується. Брунька сечовода вже не є частиною мезонеф ральної протоки, а з’єднується безпосередньо з урогенітальним синусом. З урогені тального синуса утворюється сечовий міхур і з зачатків сечовода — сечоводи. Уроге нітальний синус продовжується у сечівник каудально й у алантоїс краніально. Після облітерації алантоїса залишається урахус — фіброзний тяж, який пізніше стає меді альною пупковою зв’язкою у дорослих.
Агенезія нирок є тяжкою патологією. За відсутності нирок плід може видаляти продукти метаболізму через плаценту, але це призводить до відсутності навколо плідних вод (агідрамніону). Без амніотичної рідини легені плода не зазнають постій ного тиску, що спонукало б їх до розширення і росту. Це спричинює гіпоплазію легенів. Крім того, за відсутності амніотичної рідини плід не має можливості руха тися, що призводить до формування контрактур кінцівок плода. Спроби ввести ам ніотичну рідину в плодовий міхур при амніоцентезі виявилися безуспішними внаслі док її швидкої абсорбції. Введення постійного катетера в амніотичну порожнину несе високий ризик інфекції. Отже, на сьогодні відсутнє адекватне лікування синд рому Поттера. Але при синдромі Поттера, спричиненому вторинно внаслідок вихід ної обструкції сечового міхура, є можливість введення катетера у сечовий міхур або виконання лазерної абляції місця обструкції.
Природжений полікістоз нирок виникає з частотою 1:500–1:5000 новонароджених і успадковується за аутосомно рецесивним або аутосомно домінантним типом.
Екстрофія сечового міхура — це дефект вентральної стінки, при якому слизова оболонка сечового міхура залишається оголеною. Це рідкісна аномалія з частотою 1:100 000 новонароджених.
Існують численні аномалії нирок (сідлоподібна нирка, ектопічна нирка, подвоєння сечовода), які залишаються недіагностованими і звичайно не завдають значних про блем.
Скелетні вади
Ахондроплазія найчастіше спричинює карликовість (1:26 000 живих новонаро джених), характеризується збільшеним черепом з відносно малою середньою части ною обличчя, короткими пальцями і надмірними вигинами хребта. Ахондроплазія успадковується за аутосомно домінантним типом, 80 % випадків виникають спора дично.
Танатофорна дисплазія становить найпоширенішу летальну форму карликовості (1:20 000 живих новонароджених) і успадковується за аутосомно домінантним ти пом.
Аномалії розвитку кінцівок є різноманітними: відсутність частини кінцівки (ме ромелія), повна відсутність однієї або більше кінцівок (амелія), неправильна форма кісток (фокомелія), укорочення кінцівки (мікромелія). Класичним прикладом тера тогенного впливу є розвиток аномалій кінцівок плода, пов’язаних із прийомом ма тір’ю снодійного й антинудотного препарату талідоміду. Амніотичні перетяжки мо жуть викликати циркулярне перетискання кінцівок, пальців і навіть їх ампутацію.
Полідактилія — надкомплектні пальці кисті або стопи. Ці пальці часто не забез печені м’язами.
Природжений вивих кульшового суглоба виникає внаслідок недостатнього розвит ку кульшової западини і головки стегнової кістки. Вада частіше зустрічається у дівча ток і супроводжується слабістю капсули суглоба.
195

Акушерство і гінекологія. Том 1
Вади розвитку передньої черевної стінки плода
Вади розвитку передньої черевної стінки плода найбільш часто представлені гас трошизисом і омфалоцелє.
Гастрошизис — дефект правої параумбілікальної ділянки передньої черевної стінки, через який відбувається евісцерація тонкої кишки (рідко — інших інтраабдо мінальних органів). Гастрошизис часто є ізольованою аномалією.
Омфалоцелє — виштовхування вмісту черевної порожнини через основу пупко вого канатика, тобто наявність грижових мас, вкриті оболонками: парієтальною оче ревиною, амніоном і вартоновими драглями. Омфалоцелє нерідко асоційоване з інши ми вадами і /або хромосомними аномаліями. До 12 тиж може бути фізіологічне омфалоцелє у плода у зв’язку з екстраабдомінальним положенням кишок у цей пе ріод розвитку.
Вади розвитку м’язової системи
Частою аномалією розвитку м’язової системи є діафрагмальна грижа (1:2000 но вонароджених). У цьому разі порожнина очеревини та плевральні порожнини зали шаються сполученими між собою вздовж задньої стінки тіла і органи черевної по рожнини можуть проникати у плевральну порожнину. У 85–90 % випадків при роджена діафрагмальна грижа розвивається з лівого боку і в грудну порожнину мо жуть проникати петлі кишок, шлунок, селезінка і частина печінки. Внаслідок цього серце зміщується допереду, легені стискаються і залишаються недорозвиненими. В 75 % випадків ця вада є несумісною з життям у зв’язку з гіпоплазією і дисфункцією легенів.
Грижа стравохідного отвору може виникати внаслідок вкорочення стравоходу. При цьому верхня частина шлунка знаходиться в черевній порожнині і має місце різке звуження частини шлунка, яка проходить через діафрагму.
Вади розвитку дихальної системи і шлунково кишкового тракту
Аномалії відокремлення стравоходу і трахеї стравохідною перегородкою призво дять до атрезії стравоходу з трахеостравохідною фістулою (1:3000 новонародже них). У 90 % випадків верхня частина стравоходу закінчується сліпою кишенею, а нижній його сегмент утворює фістулу з трахеєю. Ці аномалії часто поєднуються з вадами серця (33 %), хребтовими аномаліями, анальною атрезією. Ускладненнями трахеостравохідної фістули може бути багатоводдя, оскільки амніотична рідина не може проходити до шлунка і кишок. Вміст шлунка і/або амніотична рідина можуть потрапляти до трахеї (через фістулу) і спричинювати пневмонію. Іншими аномалія ми можуть бути стеноз стравоходу, природжена грижа стравоходного отвору.
Пілоростеноз — вада розвитку шлунка внаслідок гіпертрофії циркулярної і, в меншій мірі, поздовжньої мускулатури шлунка в ділянці воріт.
Прямокишково?відхідникові атрезії та нориці (1:5000 новонароджених) утворю ються внаслідок порушень формування клоаки.
196

Розділ 6. Пренатальний скринінг...
Неперфорований анус утворюється внаслідок порушення процесу реканалізації нижньої частини відхідникового каналу.
Природжений мегаколон (хвороба Гіршпрунга) пов’язаний з відсутністю в стінці кишки парасимпатичних гангліїв.
Дивертикул Меккеля — залишок жовткової протоки у вигляді відростка клубової кишки (у 2–4 % індивідів). У ньому може міститись ектопічна тканина шлунка і підшлункової залози, що може спричинити кровотечі та навіть перфорацію.
Пренатальний скринінг
Успіх скринінгу хромосомних і природжених аномалій плода залежить від спе цифічності і чутливості скринінгових тестів.
Принципи епідеміологічних досліджень
Класичні «парні» принципи епідеміології полягають у порівнянні пар «випадок/ контроль» і «уражені/неуражені». В скринінгових тестах пари уражені/неуражені визначають як скринінг позитивні/скринінг негативні.
|
Методологія скринінгових досліджень |
|
|
|
|
||
|
Дослідні групи |
|
|
|
Скринінг позитивні |
Скринінг негативні |
|
Уражені |
a |
|
b |
Неуражені |
с |
|
d |
Чутливість (sens)= a/(a+b);
Cпецифічність (spec) = d/(c+d);
Хибнонегативні результати = b/(a+b);
Хибнопозитивні результати = c/(c+d); Позитивне прогностичне значення = a/(a+c);
Негативне прогностичне значення = d/(b+d);
Позитивне ймовірне співвідношення = sens/(1–spec) = [a/(a+b)]/[c/(c+d)];
Негативне ймовірне співвідношення = (1–sens)/spec=[b/(a+b)]/[d/(c+d)].
Чутливість тесту — це співвідношення між ураженими і скринінг позитивни ми.
Специфічність тесту — це співвідношення між неураженими і скринінг нега тивними.
Для більшої точності досліджень використовують також такі показники, як пози? тивне прогностичне значення — це відсоток уражених серед cкринінг позитивних; негативне прогностичне значення — це відсоток скринінг негативних по відношен ню до дійсно неуражених.
197

Акушерство і гінекологія. Том 1
Позитивне ймовірне співвідношення показує, як треба збільшити попередню часто ту, щоб одержати наступну частоту. Так, наприклад, якщо будь який стан зустрічаєть ся з частотою 1:100 і має позитивне ймовірне співвідношення, що дорівнює 5, то частота зустрічальності цього стану при позитивному результаті буде 5:100. Аналогіч но, негативне ймовірне співвідношення показує ймовірність негативного результату.
Пренатальний скринінг у І триместрі
У першому триместрі вагітності звичайно відбувається перший пренатальний візит і проводяться необхідні лабораторні дослідження. Перший триместр є оптималь ним часом для скринінгу, тому що багато тестів у цей період є більш чутливими і є час для встановлення діагнозу, а також, у разі необхідності, переривання вагітності.
Скринінгове ультразвукове дослідження в І триместрі виконується з метою візуа лізації та документації таких ознак:
—локалізація гестаційного мішка;
—ідентифікація ембріона;
—вимірювання тім’яно куприкового розміру і визначення гестаційного віку;
—ідентифікація серцевої активності ембріона;
—кількість плодів;
—стан матки, шийки матки і придатків матки;
—наявність, розмір і форма жовткового мішка.
Якщо діаметр гестаційного мішка перевищує 25 мм при трансабдомінальному УЗД, життєздатний ембріон може бути ідентифікований, серцева діяльність візуалі зується з 7–8 го тижня гестаційного віку. При трансвагінальному УЗД середній діа метр плідного яйця, достатній для ідентифікації, дорівнює 18 мм, серцева діяльність візуалізується з 6,5 ю тижнів гестаційного віку. Нормальний гестаційний мішок зви чайно візуалізується при УЗД при рівні β ХГЛ понад 1000 мМО/мл. При ідентифі кації серцевої активності ембріона в І триместрі частота самовільних викиднів зви чайно не перевищує 5 % і може дещо збільшуватись у віці матері > 35 років і за наявності в анамнезі самовільних викиднів. Визначення гестаційного віку ембріона в І триместрі проводять за величиною тім’яно куприкового діаметра (найбільш точ ний показник, помилка в І триместрі не перевищує 3–5 днів).
Симптом пустого плідного яйця — припинення розвитку фертилізованої яйцеклі тини. Відсутність фетального полюса в гестаційному мішку діаметром > 3 см свідчить про синдром пустого плідного яйця.
Ультразвукове дослідження товщини задньої шийної складки плода (потовщення складки) є скринінговим тестом діагностики анеуплоїдії, зокрема синдрому Дауна у плода (чутливість 60–90 %). Для підвищення точності діагностики додатково вико ристовують сироваткові тести: рівень β ?ХГЛ і плазмового протеїну А, асоційованого з вагітністю (РАРР), чутливість сироваткових тестів становить 60 %, частота хибно позитивних результатів — 5 %, а в комбінації з ультразвуковим скринінгом чут ливість зростає до 80 %.
Пренатальний скринінг у ІІ триместрі
Початковим скринінговим тестом у ІІ триместрі вагітності (15–18 тиж гестації) є дослідження рівня альфа?фетопротеїну (АФП) у сироватці крові матері, що дозво ляє виявити групу ризику щодо дефектів нервової трубки (при збільшенні рівня АФП), а також щодо розвитку синдрому Дауна.
198