

Окончание табл. 27
|
Критерий |
Дефиниция |
|
10. Иммунологические нарушения |
Анти-ДНК: антитела к нативной ДНК в повышенном титре; анти-Sm: антитела к ядерному Smith-антигену; обнаружение антифосфолипидных антител (повышенный уровень сывороточных IgG или IgМ антител к кардиолипину); обнаружение волчаночного коагулянта; ложноположительная реакция Вассермана |
|
11. Антинуклеарные антитела |
Повышение их титра, выявленное методом иммунофлюоресценции, при отсутствии приёма лекарственных средств, вызывающих волчаночноподобный синдром |
Указанные симптомы должны обязательно присутствовать в различных сочетаниях. Наличие не менее 4 критериев из них указывает на достоверность диагноза СКВ.
Лечение. Целью лечения СКВ должно быть достижение индуцированной ремиссии, которая предполагает отсутствие каких-либо клинических проявлений заболевания (при этом возможно сохранение признаков, возникших вследствие поражений того или иного органа или системы во время предшествующих обострений), отсутствие цитопенического синдрома, при иммунологическом исследовании не должны выявляться антинуклеарные и другие специфические антитела.
Общие рекомендации для больных СКВ:
исключить психоэмоциональную нагрузку;
регулярные физические упражнения позволяют уменьшить мышечную слабость;
уменьшить пребывание на солнце, использовать солнцезащитный крем;
активно лечить (и по возможности не допускать) развитие инфекции;
употреблять продукты питания с низким содержанием жира и высоким содержанием полиненасыщенных жирных кислот, кальция и витамина D;
избегать курения, злоупотребления алкоголем;
соблюдать эффективную контрацепцию в период обострения болезни и при лечении цитотоксическими лекарственными средствами (не следует принимать оральные контрацептивы с высоким содержанием эстрогенов, поскольку возможно обострение СКВ).
Основными препаратами для лечения больных СКВ являются кортикостероиды, цитостатические иммунодепрессанты (азатиоприн, циклофосфамид, хлорамбуцил), а также 4-аминохинолиновые производные (плаквенил, делагил). В последнее время получили признание методы так называемого механического очищения крови: гемосорбция, плазмаферез, лимфаферез, иммуносорбция. В качестве дополнительных средств используют антикоагулянты, симптоматичес-кие препараты, иногда НПВП. Подбор оптимальной терапии в зависимости от клинических проявлений СКВ представлен в табл. 28.
Таблица 28
Лекарственные препараты, применяемые для лечения СКВ
(цит. по Е.Л. Насонову и соавт., 2003)
|
Препарат |
Клинические проявления СКВ | ||||
|
конституциональные1 |
мышечно-скелетные |
серозит |
кожные |
тяжелые органные | |
|
НПВП |
+ |
+ |
+ |
|
|
|
ГКС локально ГКС системно: <0,5 мг/кг/сут >0,5 мг/кг/сут или пульс-терапия |
+
|
+ |
+ |
+
+ |
+ |
|
Аминохинолиновые препараты |
+ |
+ |
+ |
+ |
|
|
Азатиоприн |
+ |
+ |
+ |
+ |
+ |
|
Метотрексат |
|
+ |
|
|
|
|
Циклофосфамид |
|
|
|
|
+ |
|
Циклоспорин А |
|
+ |
+ |
+ |
+2 |
|
Аферез |
|
|
|
|
+ |
|
Внутривенный иммуноглобулин |
|
|
|
|
+3 |
|
Антикоагулянты и антиагреганты |
|
|
|
|
АФС |
|
Примечание: 1 – такие симптомы, как лихорадка, потливость, общая слабость, быстрая утомляемость, снижение работоспособности; 2 – мембранозный нефрит и тромбоцитопения; 3 – особенно тромбоцитопения. | |||||
ГКС короткого действия (преднизолон и метилпреднизолон) являются наиболее эффективными лекарственными средствами для лечения СКВ, доза этих препаратов зависит от активности заболевания:
небольшие дозы (менее 10 мг/сут) назначают при низкой активности (в случае неэффективности НПВП и аминохинолиновых препаратов);
средние дозы (менее 40 мг/сут) рекомендуют при умеренной активности (обострение артрита, полисерозита, гемолитическая анемия, тромбоцитопения и др.) в течение 2-4 недель с постепенным снижением до поддерживающей дозы;
высокие дозы (1 мг/кг/сут и более) показаны при высокой активности СКВ (риск быстрого развития необратимых поражений жизненно важных органов), длительность приема колеблется от 4 до 12 недель с последующим постепенным снижением дозы до поддерживающей (5-10 мг/сут) под тщательным клинико-лабораторным контролем.
Показаниями к применению цитостатических иммунодепрессантов у больных СКВ являются: 1) активный волчаночный нефрит; 2) высокая общая активность болезни и резистентность к ГКС или появление побочных действий этих препаратов; 3) необходимость уменьшить поддерживающую дозу преднизолона, если она превышает 15-20 мг/сут. Существуют различные схемы комбинированного лечения: азатиоприн или циклофосфамид внутрь в среднем в дозе 2-2,5 мг/кг/сут, хлорбутин (хлорамбуцил, лейкеран) по 0,2-0,4 мг/кг/сут в сочетании с низкими (25 мг) и средними (40 мг) дозами преднизолона (М.М. Иванова, 2004).
Основная эмпирическая схема лечения циклофосфамидом состоит в применении препарата внутривенно до достижения за 3-4 недели суммарной дозы 2000 мг (схемы индивидуальны: по 500 мг 1 раз в неделю или по 200 мг через день 10 раз), после чего назначают по 200 мг в неделю (внутривенно или внутримышечно) в течение 2-5 лет или по 1000 мг в месяц внутривенно при интенсивном лечении в течение 6-12 месяцев.
Азатиоприн назначают по 100-150 мг в день, обычно при стойком кожно-суставном синдроме, люпус-нефрите. При стойком суставном синдроме и при некоторых формах поражения ЦНС может оказаться эффективным метотрексат. За последние годы получен опыт применения циклоспорина А при активном люпус-нефрите в дозе 3-5 мг/кг/сут на протяжении 7-20 месяцев в сочетании с 16-20 мг метипреда в день.
Интенсивная терапия, то есть назначение сверхвысоких (ударных) доз ГКС и цитостатиков для подавления активности иммунного воспаления, впервые в нашей стране при СКВ была применена 20 лет назад (М.М. Иванова и соавт., 1983) и показала высокую эффективность при тяжелом течении болезни.
Основные показания к применению интенсивной терапии:
активный люпус-нефрит (особенно с нефротическим синдромом, артериальной гипертензией, быстрым повышением уровня креатинина);
острое тяжелое поражение ЦНС (менингоэнцефалит, энцефаломиелополирадикулоневрит, поперечный миелит);
гематологический криз, выраженная тромбоцитопения;
язвенно-некротический кожный васкулит;
легочный васкулит;
высокая активность болезни, резистентная к предшествующей, считающейся ранее адекватной, терапии (М.М. Иванова и соавт., 2004).
Основной метод интенсивной терапии СКВ – пульс-терапия ГКС, которую проводят метилпреднизолоном в дозе 1000 мг/сут внутривенно 3 дня подряд.
Ниже приведены наиболее распространенные методики интенсивной терапии (М.М. Иванова и соавт., 2004):
классическая пульс-терапия метилпреднизолоном: по 1000 мг/сут внутривенно капельно в течение 3 последовательных дней (3000 мг на курс);
ежедневное внутривенное введение метилпреднизолона в уменьшенных дозах (250-500 мг/сут) до достижения суммарной дозы около 3000 мг на курс;
ежемесячное внутривенное введение 1000 мг метилпреднизолона в течение 6-12 месяцев;
комбинированная пульс-терапия: внутривенное введение по 1000 мг метилпреднизолона 3 дня подряд и по 1000 мг циклофосфана в 1-й или 2-й день (препараты вводят последовательно);
ежемесячное внутривенное введение 1000 мг метилпреднизолона и 1000 мг циклофосфана в течение 12 месяцев.
Следует отметить, что быстро снижать пероральную суточную дозу преднизолона непосредственно после проведения пульс-тера-пии ГКС не рекомендуется. При высокой активности болезни после пульс-терапии, которую применяют обычно утром, на вечер оставляют ¼ суточной дозы per os, так как метилпреднизолон, введенный утром внутривенно, через 4-7 ч в крови уже не определяется и может развиться синдром отмены.
К дополнительным методам патогенетической терапии СКВ следует отнести экстракорпоральные методы. Применение плазмафереза и гемосорбции основано на возможности удаления из крови биологически активных веществ: медиаторов воспаления, циркулирующих иммунных комплексов, криопреципитинов, различных антител и др. Полагают, что механическое очищение помогает на некоторое время «разгрузить» систему мононуклеаров, стимулируя таким образом эндогенный фагоцитоз новых комплексов, что в итоге уменьшает степень органных повреждений.
Одним из наиболее активных методов «программного» лечения является синхронная интенсивная терапия: проведение курса плазмафереза (3-6 процедур) с последующей комбинированной пульс-терапией ГКС и циклофосфаном. При этом обычно сразу после первой процедуры плазмафереза последовательно внутривенно капельно вводят 1000 мг метилпреднизолона и 1000 мг циклофосфана, а после повторных сеансов плазмафереза при курсовом лечении ежемесячно вводят внутривенно только метилпреднизолон в дозе 500-1000 мг в течение 12 месяцев и более (С.К. Соловьев и соавт., 1998).
Смертность при СКВ в три раза выше, чем в популяции. К факторам, ассоциирующимся с неблагоприятным прогнозом, относятся: поражение почек (особенно диффузный пролиферативный гломерулонефрит), артериальная гипертензия, мужской пол, дебют заболевания в пожилом или детском возрасте, антифосфолипидный синдром, высокая активность заболевания, присоединение интеркуррентной инфекции, осложнения лекарственной терапии.
Дерматомиозит(ДМ) – системное заболевание соединительной ткани, характеризующееся прогрессирующим воспалительным поражением скелетной мускулатуры с нарушением двигательных функций, типичными изменениями кожи и системными проявлениями (синдром Рейно, поражение легких, сердца и др.) (Н.Г. Гусева, 2004). У 25-30 % больных кожный синдром отсутствует, в этом случае используется термин «полимиозит» (ПМ). Согласно современной международной классификации, ДМ относится к группе системных заболеваний соединительной ткани и является характерным представителем аутоиммунных воспалительных миопатий.
Классификация. ДМ является одной из форм воспалительных миопатий, которые подразделяются на три группы: идиопатические, вызываемые инфекциями и вызываемые лекарственными средствами и токсинами (R.L.Woltman, 1994). ДМ относится к идиопатическим воспалительным миопатиям. Наиболее известной и широко используемой является приводимая ниже классификация ДМA.BohanиY.Peter(1975).
Классификация ДМ (ПМ)
Первичный (идиопатический) полимиозит.
Первичный (идиопатический) дерматомиозит.
Дерматомиозит (или полимиозит) в сочетании с неоплазмой.
Детский дерматомиозит (или полимиозит) в сочетании с васкулитом.
Полимиозит или дерматомиозит в сочетании с другими диффузными заболеваниями соединительной ткани.
Выделяют также острое, подострое, затяжное и хроническое течение ДМ и ПМ. Острое течение характеризуется лихорадкой, генерализованным поражением поперечнополосатой мускулатуры, вплоть до полной обездвиженности, прогрессирующей дисфагией, висцеритами, эритематозным поражением кожи, возникшими в течение первых 6 месяцев с момента манифестации болезни. Для подострого варианта течения заболевания характерно более медленное нарастание симптомов, однако уже через 1-2 года наблюдается развернутая клиническая картина ДМ (ПМ) с тяжелыми поражениями мышц, кожи и внутренних органов. При хроническом течении преобладают процессы атрофии и склероза мышечной ткани, поражение кожных покровов в виде гиперпигментации и гиперкератоза, висцеральные проявления развиваются реже.
По степени клинико-лабораторной активности принято выделять минимальную (I), умеренную (II), высокую (III) активность и ремиссию.
Частота ДМ (ПМ) составляет 5-10 новых случаев на 1 млн населения в год. Заболевание чаще поражает женщин, соотношение по полу среди взрослых больных (женщин и мужчин), по данным большинства авторов, составляет 2:1 и более. Отмечено два возрастных пика болезни: 11-17 и 35-60 лет.
ЭтиологияДМ до конца не изучена. Не исключается вирусная природа заболевания (пикорнавирусы, вирусы Коксаки, краснухи, герпеса), а также участие бактериальных, паразитарных (риккетсиоз, шистоматоз, трихинеллез и др.) инфекций. Провоцирующими факторами могут быть переохлаждение, инсоляция, избыточная физическая нагрузка и др. Неопластический ДМ составляет 20-30 % всех случаев заболевания, особенно в старшей возрастной группе (свыше 50 лет).
ПатогенезДМ и ПМ аутоиммунный. Он характеризуется синтезом широкого спектра аутоантител, направленных против цитоплазматических белков и рибонуклеиновых кислот (РНК), принимающих участие в синтезе белка. Эти антитела редко выявляются при других аутоиммунных заболеваниях и рассматриваются как миозитспецифические, условно подразделяемые на 4 группы:
антитела к аминоацилсинтетазам транспортной РНК (тРНК);
антитела, реагирующие с частицами сигнального распознавания и блокирующие перенос вновь синтезированных белковых молекул к эндоплазматический сети;
антитела, реагирующие с белково-ядерным комплексом;
антитела, реагирующие с фактором элонгации i-альфа, который обеспечивает перемещение аминоацил-тРНК к рибосомам и движение вдоль полисомы.
Миозитспецифические антитела при ДМ и ПМ обнаруживаются в 40 % случаев, при этом каждый больной имеет только один тип антител. Однако наряду с миозитспецифическими антителами в сыворотке крови у этих пациентов могут присутствовать и другие, неспецифические типы аутоантител, например антитела к миозину, тиреоглобулину, ревматоидный фактор и др. Схематически патогенез ДМ (ПМ) представлен на рис. 16.
К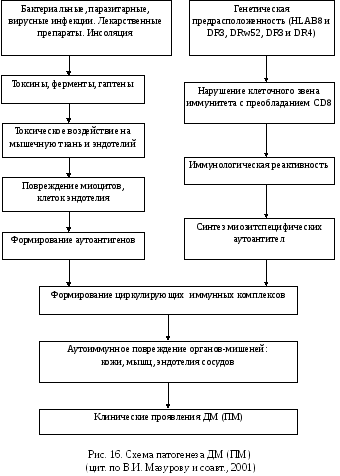 линическая
картина. Начало заболевания может
быть острым, но чаще симптоматика
развивается постепенно, характеризуясь
преимущественно кожными и мышечными
проявлениями: отек и гиперемия в
периорбитальной области, на открытах
частях тела, миалгии, нарастающая
мышечная слабость, иногда артралгии,
субфебрилитет. Частота (%) основных
клинических проявлений ДМ, по данным
Е.М. Тареева и А.П. Соловьевой (1985),
представлена ниже.
линическая
картина. Начало заболевания может
быть острым, но чаще симптоматика
развивается постепенно, характеризуясь
преимущественно кожными и мышечными
проявлениями: отек и гиперемия в
периорбитальной области, на открытах
частях тела, миалгии, нарастающая
мышечная слабость, иногда артралгии,
субфебрилитет. Частота (%) основных
клинических проявлений ДМ, по данным
Е.М. Тареева и А.П. Соловьевой (1985),
представлена ниже.
Повышение температуры тела – 67,7.
Поражение кожи – 100.
Эритема – 100.
Периорбитальный отек – 95.
Капилляриты – 66.
Отек – 70.
Синдром Рейно – 0.
Генерализованное поражение скелетных мышц – 100.
Слабость – 100.
Миалгия – 97.
Контрактуры – 7.
Кальциноз – 9.
Дисфагия – 75.
Поражение слизистых оболочек – 52,5.
Артрит/артралгия – 7/27,7.
Поражение сердца – 33.
Поражение миокарда – 33.
Поражение эндокарда – 0,5.
Поражение перикарда – 0.
Адгезивный плеврит – 6.
Нефрит – 21,5.
Гепатомегалия (жировая дистрофия) – 39.
Поражение кожипри ДМ полиморфно: преобладают эритема, отек и дерматит преимущественно на открытых участках тела. Наблюдаются папуллезные, буллезные, петехиальные высыпания, телеангиэктазии, очаги пигментации и депигментации, гиперкератоза. Характерны периорбитальный отек и эритема, имеющая своеобразный лиловый оттенок (симптом «очков»), играющий важную диагностическую роль при постановке диагноза. Выраженная эритема чаще локализуется на лице, шее, в зоне декольте, над проксимальными межфаланговыми и пястно-фаланговыми суставами (симптом Готтрона), на наружной поверхности предплечья и плеча, передней поверхности бедер и голеней. Нередко наблюдаются трофические нарушения в виде сухости кожи, ломкости ногтей, выпадения волос и др. У каждого второго больного ДМ отмечаются конъюнктивит, стоматит, отек зева, голосовых связок.
Поражение скелетных мышцявляется ведущим признаком ПМ и ДМ. Характерно развитие тяжелого, нередко некротического миозита с преимущественным поражением мышц проксимальных отделов конечностей, плечевого и тазового пояса, шеи, спины, глотки, верхних отделов пищевода, сфинктеров.
Клинически отмечают боль в мышцах, плотность или тестоватый характер пораженных мышц, увеличение их в объеме, болезненность при пальпации. Доминирующий признак – неуклонно прогрессирующая мышечная слабость, выражающаяся в значительном ограничении активных движений пациента, что не позволяет им самостоятельно встать, сесть, поднять ногу на ступеньку (симптом «автобуса»), удержать какой-либо предмет в руке, причесаться, одеться (симптом «рубашки»). При поражении мышц шеи и спины больные не могут самостоятельно приподнять голову от подушки (симптом «подушки») или удержать ее сидя (голова падает на грудь). Практически затруднены все движения, связанные с участием проксимальных мышц конечностей, но в то же время в дистальных отделах (в кистях и стопах) сохраняется полный объем движений.
Вовлечение в процесс глоточных мышц ведет к дисфагии, возможна аспирация пищи в трахею. В отличие от дисфагии, наблюдающейся при системной склеродермии, у больных ДМ затруднено глотание как твердой, так и жидкой пищи (поперхивание при приеме пищи). Поражение межреберных мышц и диафрагмы способствует развитию пневмонических осложнений, что может служить причиной летального исхода. В случае поражения мышц гортани отмечается дисфония, охриплость голоса, вплоть до афонии.
Тяжесть состояния и инвалидизация больных ДМ обусловлены также нередким последующим развитием сухожильно-мышечных контрактур, атрофией и кальцинозом ранее пораженных групп мышц. Кальцинируются обычно участки фасции, подкожной клетчатки, прилежащие к пораженным мышцам, при этом массивные участки кальциноза могут быть резко болезненны.
С целью стандартизации оценки состояния больного по ведущему признаку заболевания – мышечной слабости – предложена градация ее выраженности с использованием 6 степеней (табл. 29) по A.RoseиJ.Walton(1966).
Таблица 29
Градация мышечной слабости при ДМ (ПМ)
|
Степень |
Выраженность мышечной слабости |
|
I |
Нет нарушений в момент осмотра |
|
II |
Нет нарушений при осмотре, но имеются небольшая слабость и снижение толерантности к физической нагрузке |
|
III |
Небольшая атрофия одной или более мышечных групп без функциональных нарушений |
|
IV |
Нарушения функции: неспособность бегать, но сохранение возможности идти по лестнице без поддержки рук |
|
V |
Выраженная мышечная слабость, лордоз, неспособность идти по лестнице (спускаться) или подниматься со стула без использования рук (или помощи) |
|
VI |
Неспособность встать без помощи окружающих |
Суставной синдромне является ведущим в клинике ДМ и ПМ; характерны артралгии, поражение периартикулярных тканей, артриты возникают редко. Чаще в процесс вовлекаются локтевые, плечевые, коленные суставы и кисти. Нарушение функции суставов и контрактуры связаны с поражением мышц.
Поражение внутренних органов обычно встречается у большинства больных ДМ, но не превалирует в картине болезни, как при ССД и СКВ.
При поражении сердцаобычно наблюдаются нарушения ритма (тахикардия и аритмия), формирование застойной сердечной недостаточности, связанные с развитием миокардита или миокардиофиброза. Описаны случаи дилатационной кардиомиопатии и констриктивного перикардита.
Поражение легкиху больных ДМ обусловлено рядом факторов и включает участие мышечного синдрома (гиповентиляция), инфекционных агентов, аспирацию при нарушении глотания, развитие интерстициальной пневмонии и фиброзирующего альвеолита. Легочный фиброз, обусловленный интерстициальным поражением ткани легких, легочным васкулитом и развитием септально-альвеолярного склероза, отмечается у 5-10 % больных. Он характеризуется нарастающей инспираторной одышкой, сухим кашлем, звучными хрипами в нижних отделах легких, прогрессирующей дыхательной недостаточностью.
Поражение почекпри ДМ (ПМ) встречается относительно редко. При остром течении тяжелая персистирующая миоглобинурия может привести к развитию почечной недостаточности. По данным Л.И. Исаевой и соавт. (1978), среди больных ДМ у 41,5 % отмечается транзиторная протеинурия с микрогематурией и цилиндрурией.
Лабораторные исследования характеризуют в основном общую активность ДМ и только появление креатина в моче и повышение в крови уровня креатинфосфокиназы (КФК), аминотрасфераз и альдолазы свидетельствует непосредственно об остроте и распространенности поражения мышц. У некоторых больных ДМ наблюдаются умеренная анемия, лейкоцитоз, реже – лейкопения, эозинофилия, увеличение СОЭ, повышение уровня СРБ, γ-глобулинов, фибриногена, сиаловых кислот, серомукоида. У большинства пациентов наблюдается повышение уровня миоглобина в сыворотке крови, реже – миоглобинурия. Часты иммунные нарушения – обнаружение различных антиядерных и миозитспецифических антител, иногда РФ и волчаночного антикоагулянта (обычно в низких титрах), иммунных комплексов и др.
Из биохимических тестов наиболее характерно повышение сывороточного уровня мышечных ферментов, отражающих выраженность поражения мышц, в первую очередь – КФК и в меньшей степени альдолазы и аминотрансфераз. При этом содержание креатинфосфокиназы может превышать нормальный уровень в 80 раз, в среднем же увеличивается в 5-10 раз.
С помощью электромиографии выявляют снижение амплитуды и укорочение продолжительности биопотенциалов пораженных мышц, полифазность, иногда – спонтанную активность типа фибрилляции, псевдомиотонические нарушения и др. Для ДМ (ПМ) характерна следующая триада электромиографических изменений:
спонтанная фибрилляция и положительные потенциалы, как при денервации мышц;
появляющийся при произвольном сокращении мышцы полиморфный комплекс потенциалов, амплитуда которых значительно меньше, чем в норме;
залпы высокочастотных потенцалов действия («псевдомиотония») после механического раздражения мышцы.
Данные электромиографии не являются строго специфичными для ДМ (ПМ), но в сочетании с клинической картиной и другими исследованиями достаточно широко используются для диагностики этого заболевания. Предпочтение отдают игольчатой электромиографии. Подчеркивается значение отдельных признаков как для подтверждения собственно поражения мышц, так и для уточнения его характера. Так, полифазность потенциалов наряду с другими миогенными признаками является аргументом в пользу «миозитного» процесса, при этом число бифазных комплексов преобладает над трехфазными.
При биопсии мышц, которую проводят в области поражения (мышцы плеча, бедра и др.), обычно обнаруживают выраженные изменения воспалительного и дегенеративного характера: клеточную инфильтрацию с преобладанием лимфоцитов, гистиоцитов и плазматических клеток, некроз мышечных волокон с потерей поперечной исчерченности, дегенеративными изменениями, фагоцитозом и элементами регенерации. Как правило, отмечается сосудистая патология в виде сегментарных пролиферативных васкулитов, утолщения интимы и склероза стенки мелких сосудов, сужения просвета, тромбозы. Регенерация характеризуется наличием малых волокон с большими ядрами, везикулярными и нуклеолярными структурами.
При хроническом процессе увеличивается количество разнокалиберных волокон, возрастает число ядер внутри волокон, эндо- и перимизиальный фиброз. Необходимо отметить, что атрофия мышечных волокон отчетливо преобладает над гипертрофией.
Критерии диагностики. Официально принятых международных критериев ДМ нет, но в клинической практике наиболее часто используются диагностические критерии, разработанныеA.BohanиJ.Peterв 1975 году.
Критерии диагноза ПМ и ДМ (A.Bohan,J.Peter, 1975)
Симметричная слабость проксимальных мышц плечевого и тазового пояса и передних сгибателей шеи, прогрессирующая в течение нескольких недель (или месяцев) в сочетании или при отсутствии дисфагии или поражения дыхательной мускулатуры.
При гистологическом исследовании мышц – признаки некроза мышечных фибрилл IиIIтипов, фагоцитоз, регенерация с базофилией, крупные ядра и ядрышки в сарколемме, перифасциальная атрофия, вариабельность размера миофибрилл, воспалительный экссудат (по данным биопсии).
Повышение сывороточной концентрации мышечных (миоспецифических) ферментов (КФК, альдолаза, аспарагиновая и аланиновая аминотрансферазы, ЛДГ), а также миоглобина в сыворотке и моче, креатинурия.
Электромиографические изменения (приведенная выше электромиографическая триада): короткие, мелкие полифазные моторные единицы, фибрилляции и т.д.
Дерматологические проявления, включающие гелиотропную окраску кожи век с периорбитальным отеком; чешуйчатый эритематозный дерматит на тыльной поверхности кистей, особенно над пястно-фаланговыми и проксимальными межфаланговыми суставами (симптом Готтрона) и поражение кожи над коленными, локтевыми суставами, лица, шеи, верхней половины грудной клетки.
При наличии первых четырех критериев можно говорить об «определенном» диагнозе ПМ, а наличие пяти критериев позволяет диагностировать «определенный» ДМ.
Эти критерии были пересмотрены и дополнены К. Tahimotoи соавт. в 1995 году.
Изменения кожи:
гелиотропная кожная сыпь (светло-фиолетовая эритема с отеком верхних век);
симптом Готтрона (коллоидные пятна на тыльной стороне суставов пальцев кисти);
эритема на тыльной стороне суставов конечностей: слегка возвышающаяся, незначительно щелушащаяся, бледно-фио-летовая эритема над локтевыми и коленными суставами.
Слабость проксимальных мышц (верхних или нижних конечностей и туловища).
Повышенный уровень сывороточной КФК или альдолазы.
Боль в мышцах при давлении или спонтанная.
Патологические изменения электромиограммы (короткие многофазовые потенциалы, фибрилляции и псведомиотонические разряды).
Обнаружение анти-Jo-1 (гистадил-тРНК-синтетаза) антител.
Недеструктивный артрит или артралгии.
Признаки системного воспаления (лихорадка 37˚С, увеличение СРБ или СОЭ выше 20 мм/ч).
Миозит, обнаруженный в биоптате мышцы (инфильтрация скелетной мышцы с воспалительными клетками и фокальной или экстенсивной дегенерацией мышечных волокон вплоть до некроза и регенеративных процессов с неравномерным замещением волокон фиброзом).
При наличии хотя бы одного кожного изменения и как минимум 4 критериев из последующих ДМ весьма вероятен (чувствительность 94,1 % и специфичность 90,3 %).
При наличии как минимум 4 критериев со 2-го по 9-й весьма вероятен ПМ (чувствительность 98,9 % и специфичность ПМ, по сравнению со всеми контрольными заболеваниями, 95,2 %).
Несмотря на характерную клиническую картину болезни, диагностика ее, особенно в дебюте, представляет большие затруднения. При кожной симптоматике чаще всего преобладают ошибочные «дерматологические» диагнозы, а при мышечной – «неврологические». Важно не только установить диагноз ДМ (ПМ), но и определить его клиническую форму, провести дифференциальную диагностику первичного (идиопатического) и вторичного (опухолевого) ДМ (ПМ), исключить другие состояния, сопровождающиеся распространенным поражением скелетных мышц.
Трудность дифференциальной диагностики подтверждается наличием большого количества заболеваний с поражением мышц другого генеза (W.Bradley, 1981).
Денервационные состояния: спинальная мышечная атрофия, боковой амиотрофический склероз.
Поражение нейромышечного соединения: синдром Итона–Ламберата,myastheniagravis.
Генетическая мышечная дистрофия: лицелопаточно-плечевая (бедренная), дистальная, окулярная и др.
Миотонические заболевания: миотоническая дистрофия, врожденная миотония.
Липидные болезни (нарушения липидного обмена): карнитин-дефицит, карнитин-пальмитин-трансферазный дефицит и др.
Гликогеновые болезни: мальтаза-дефицитная с началом во взрослом состоянии, болезнь Мак-Ардля (мышечная форма гликогеноза).
Периодический паралич.
Оссифицирующий миозит– генерализованный и локальный.
Эндокринные миопатии: гипотиреоз, тиреотоксикоз, акромегалия, болезнь Кушинга, болезнь Аддисона, гиперпаратиреоз, гипопаратиреоз, миопатия, обусловленная дефицитом витаминаD, гипокалиемия, гипокальциемия.
Метаболические миопатии: уремия, печеночная недостаточность.
Токсические миопатии: острый и хронический алкоголизм; лекарственная миопатия (включаяD-пеницилламин, хлорохин, эмитин и др.).
Миопатии нарушения питания: дефицит витамина Е, нарушения всасывания и др.
Карциноматозная миопатия: карциноматозная кахексия.
Проксимальная нейропатия: синдром Гийена–Барре, острая интермиттирующая порфирия, диабетическая хроническая плексопатия, хроническая аутоиммунная полинейропатия.
Микроэмболизация атеромой или карциномой.
Ревматическая полимиалгия.
Другие заболевания соединительной ткани: РА, ССД, СКВ, узелковый полиартериит.
Инфекционные заболевания: острые вирусные, мононуклеоз, риккетсиозы, Коксаки-вирусные, краснуха и вакцинация против нее, острое бактериальное поражение.
Паразитарные заболевания, включая токсоплазмоз, трихинеллез, шистосомоз, цистицеркоз и др.
Септический миозит, включая стафилококковый, стрептококковый, лепрозный и др.
При установленном диагнозе ДМ (ПМ) жизненно важна для больного дифференциация первичного «идиопатического» и вторичного (опухолевого) ДМ, определяющая тактику лечения и прогноз. Миозит, ассоциирующийся с опухолями, составляет, по данным различных авторов, от 6,7 до 34 % среди всех случаев воспалительных миопатий. Было показано, что на фоне злокачественных новобразований более часто развивается ДМ, чем ПМ. Частота заболевания одинакова у мужчин и женщин (1:1). Практически любая злокачественная опухоль, включая заболевания крови, может ассоциироваться с развитием вторичного ДМ (ПМ); чаще наблюдаются неоплазмы органов половой сферы, легких, ЖКТ, почек. Связь ДМ с опухолью четко аргументируется случаями излечения после удаления опухоли и, наоборот, рецидивом или прогрессированием его при наличии метастазов (Н.Г. Гусева, 2004). Нормальные значения КФК у больных с типичными проявлениями миозита могут указывать на его связь с неопластическим процессом. По данным Н.Г. Гусевой и соавт. (2004), к «факторам риска» выявления злокачественных опухолей, помимо возраста и пола, следует отнести атипичное течение и рефрактерность ДМ (ПМ) к проводимой кортикостероидной терапии, и наоборот – наличие миозитспецифических антител, которые практически не наблюдаются при опухолевом ДМ, или сочетание ДМ с другими заболеваниями соединительной ткани, значительно снижают вероятность выявления неоплазм. Авторы рекомендуют для всех больных ДМ, особенно при наличии факторов риска, проводить скринирующие исследования на неоплазму: 1) маммографию; 2) исследование органов малого таза у женщин и предстательной железы у мужчин; 3) эндоскопическое исследование органов грудной и брюшной полости.
Лечение. Основой в лечении больных ДМ (ПМ) признана кортикостероидная терапия. ГКС короткого действия (преднизолон, метилпреднизолон) остаются единственной группой препаратов, эффективность которых доказана в контролируемых исследованиях. По данным некоторых авторов, полный или частичный ответ на ГКС в адекватной дозе (а минимальная эффективная доза должна составлять не менее 1 мг/кг массы тела) удается достичь только у 75-90 % больных. В первые недели препарат следует назначать в несколько приемов, а затем переводить больного на однократный прием всей дозы в утренние часы. К сожалению, улучшение состояния пациентов с ДМ (ПМ) при лечении высокими дозами ГКС происходит не так быстро, как при других ревматических заболеваниях (в среднем через 2-4 месяца).
Лечение кортикостероидами улучшает состояние у подавляющего большинства больных ДМ, радикально – при первичном ДМ и частично – при вторичном (паранеопластическом), где решающим отстается эффективное оперативное вмешательство и иные виды терапии. Оказывая противовоспалительное и иммунодепрессивное действие, ГКС в больших дозах способны подавить воспалительный и аутоиммунный процессы в мышечной ткани, препятствуя развитию некроза и последующих фиброзно-атрофических и дистрофических изменений. Эффективность лечения контролируют с помощью клинических и лабораторных тестов, включая исследование уровня КФК. Большие дозы ГКС принимаются больными длительно, нередко на протяжении 0,5-1 года. Уменьшение дозы преднизолона проводится постепенно с обязательным соблюдением общего правила: чем меньше доза, тем больше интервал перед следующим ступенеобразным снижением ее. Так, при дозе 100-80 мг преднизолона в день возможно снижение ее по ½ таблетки каждые 3-5 дней, при 70-40 мг – по ½ таблетки в 5-10 дней или по ¼ таблетки в 3-4 дня, при 30 мг – по ¼ таблетки в 7-10 дней, при 20 мг – по ¼ таблетки в 3 недели; далее еще медленнее (В.И. Мазуров и соавт., 2001).
Длительная терапия высокими дозами ГКС всегда сопряжена для больного с риском развития таких осложнений, как синдром Иценко–Кушинга (ожирение, стрии и др.), остеопороз и стероидная спондилопатия, стероидный диабет, желудочно-кишечные кровотечения, инфекционные осложнения, кардиомиопатии, нарушение психики, что требует проведения симптоматической терапии, а иногда снижения дозы и комбинации с другими препаратами (иммунодепрессанты).
Дополнительный прием препаратов кальция и витамина D, анаболических стероидов может замедлить развитие остеопороза. В период лечения высокими дозами ГКС показаны препараты калия, антисекреторы и антациды; при задержке жидкости – калийсберегающие диуретики, при склонности к гипертензии – антигипертензивная терапия. При наличии очагов инфекции рекомендуют подключать антибиотики и антимикотические средства.
При высокой клинико-лабораторной активности процесса, наличии висцеральных проявлений, прогрессирующей миопатии проводится пульс-терапия мегадозами метилпреднизолона по схеме, описанной выше в разделе «лечение СКВ».
Альтернативными лекарственными средствами в лечении ДМ (ПМ) могут служить цитостатические препараты, которые чаще всего применяются в сочетании с ГКС. В комбинированной терапии роль иммунодепрессантов сводится к так называемому «стероидсберегающему» действию (возможность достигнуть клинический эффект в ответ на меньшую дозу ГКС). При ДМ (ПМ) применяют метотрексат, азатиоприн, циклофосфамид, хлорбутин. Показаниями для назначения цитостатиков являются:
резистентные к максимально высоким дозам ГКС формы ДМ (ПМ);
наличие сопутствующих заболеваний или побочных эффектов, ограничивающих возможность адекватной терапии ГКС;
принадлежность больных ДМ (ПМ) к определенным клинико-иммунологическим подтипам, особенностью которых является плохой ответ на ГКС.
Ниже приводятся дозы цитостатических препаратов, используемых в лечении ДМ (ПМ):
метотрексат внутрь или внутривенно 7,5-25 мг/нед.;
азатиоприн (максимальный клинико-лабораторный эффект проявляется только через 6-9 месяцев) внутрь 2-3 мг/кг/сут (100-200 мг/сут);
циклофосфамид по 2 мг/кг/сут;
циклоспорин А внутрь 2,5-5,0 мг/кг/сут;
гидроксихлорохин (только при кожных проявлениях ДМ) 200 мг/сут.
Внутривенное введение высоких доз иммуноглобулина многие клиницисты рассматривают как один из наиболее перспективных методов лечения аутоиммунных заболеваний, в том числе ДМ (ПМ). Применяют 2 схемы введения иммуноглобулина: по 1 мг/кг массы тела в течение 2 дней и по 0,5 мг/кг в течение 4 дней ежемесячно (общая продолжительность 3-4 месяца).
Плазмаферез и лимфоцитаферез показан пациентам с тяжелым ДМ (ПМ), резистентным к другим методам терапии; лечение обязательно необходимо сочетать с применением ГКС и цитостатиков.
Важное значение имеют реабилитационные мероприятия, которые следует проводить дифференцированно в зависимости от стадии заболевания. В острой фазе показаны пассивные упражнения и напряжение мышц, в стадии ремиссии – изометрические, а затем изотонические упражнения. При преобладании процессов атрофии и фиброза мышц с развитием контрактур в терапевтический комплекс включаются лечебная гимнастика, массаж (но не глубокий и не травмирующий ткани), физиотерапевтические процедуры (парафин, электрофорез прозерина, гиалуронидазы и др.).
Внедрение в клиническую практику ГКС существенно увеличило выживаемость больных ДМ (ПМ), которая в целом по группе (за исключением пациентов с миозитом, сопровождающимся злокачественными новообразованиями) составляет 90 % через 5 лет после постановки диагноза.