

Васкулиты с преимущественным поражением сосудов мелкого калибра:
Микроскопический полиангиит.
Геморрагический васкулит.
Кожный лейкоцитокластический васкулит.
Смешанные состояния:
Облитерирующий тромбангиит (болезнь Винивартера–Бюргера).
Болезнь Кавасаки.
Как видно из вышеприведенной классификации, в патологический процесс могут вовлекаться сосуды разных сосудистых регионов и любого калибра– от аорты до капилляров, что и обусловливает многоликость клинических проявлений. Так, при поражении сосудов крупного калибра ведущим синдромом является ишемия наряду с общевоспалительными реакциями в виде лихорадки, похудания, общей слабости, при этом в зависимости от локализации воспалительного процесса в сосудистой сети развиваются нарушения зрения вплоть до слепоты, перемежающаяся хромота, интенсивные головные боли, миалгии. Некротизирующие васкулиты протекают достаточно тяжело вследствие вовлечения в патологический процесс сосудов среднего и мелкого калибра, прогноз этих заболеваний определяется степенью поражения жизненно важных органов (почки, сердце, легкие, ЦНС) и быстротой развития полиорганной недостаточности. В случае же гиперергических васкулитов (аллергические ангииты), когда поражаются преимущественно мелкие сосуды, в клинической картине преобладает кожный синдром (кожная пурпура).
СВ чаще встречаются у мужчин, чем у женщин, могут развиваться в любом возрасте, но чаще у лиц среднего возраста. Пик заболеваемости нередко приходится на зиму и весну, что, вероятно, может быть связано с сезонной активацией жизненного цикла микроорганизмов.
Этиологияпервичных СВ по-прежнему остается неизвестной. Не исключается роль вирусной инфекции (в частности, вирус гепатита В, цитомегаловирус, вирус иммунодефицита человека,herpes simplex), а в последние годы вновь появилось немалое число работ о роли бактериальной и паразитарной инфекции как триггерных факторов в развитии СВ. Необходимо отметить, что наличие несанированных очагов хронической инфекции может привести к рецидивированию заболевания и развитию вторичных инфекционных осложнений, что усугубляет прогноз у больного с СВ. Кроме того, нередко в роли причинного фактора могут выступать лекарства. В настоящее время известно более 100 препаратов, прием которых приводит к развитию васкулита, среди них сульфаниламиды, антибиотики, препараты йода, рентгеноконтрастные вещества, витамины группы В, анальгетики, туберкулостатики и др. Нельзя исключить и наследственную предрасположенность, что обусловлено дефектом иммунного ответа и измененной реактивностью стенки сосуда.
Выделяют несколько механизмов повреждения сосудистой стенки:
непосредственное (прямое) влияние химической субстанции или микроорганизма без участия иммунопатологических реакций;
образование органоспецифических антител к различным структурам макроорганизма (например, к базальной мембране клубочков почек, эндотелию сосудов);
циркуляция в сосудистом русле иммунных комплексов, при этом осаждение их на стенке сосуда вызывает воспаление;
наличие АНЦА (реагирующие с различными ферментами цитоплазмы нейтрофилов), обладающих способностью к перекрестному взаимодействию с эндотелием, то есть проявляют активность антиэндотелиальных антител.
По современным представлениям, в развитии СВ одновременно принимают участие несколько иммунных, а возможно и неиммунных, патологических механизмов (F.C.Breedveldetal., 1996;P.A.Bacon, 1996). Большое значение отводится активации клеточного иммунитета, характеризующейся в некоторых случаях преобладаниемTh1-типа (Т-хелперы 1-го типа) иммунного ответа (с гиперпродукцией ИЛ-2 и ИФ-γ), с увеличением выработки «провоспалительных» цитокинов (таких как ИЛ-1 и ФНО-α), инфильтрацией Т-лимфоцитами и макрофагами стенки сосуда, образованием гранулем. При этом одним из этапов реализации эффекторного иммунного воспаления является продукция активированными макрофагами и нейтрофилами гистодеструктивных (протеолитических) ферментов и активных форм кислорода. Не менее важное звено патогенеза васкулитов связано с активацией гуморального иммунитета, проявляющейся выработкой аутоантител (например, антитела к компонентам цитоплазмы нейтрофилов, эндотелию сосудов, фосфолипидам базальной мембраны), образованием иммунных комплексов. В развитии СВ немаловажная роль отводится активации самого сосудистого эндотелия, увеличению экспрессии на его поверхности молекул адгезии, нарушению нормальных механизмов апоптоза эндотелиоцитов и взаимодействию между ними и лейкоцитами; было показано, что все эти факторы могут вызывать увеличение связывающей активности эндотелия, потерю им поверхностных компонентов с антикоагулянтной активностью и стимулировать синтез и/или высвобождение прокоагулянтных субстанций (Е.Л. Насонов, А.А. Баранов, Н.П. Шилкина, 1999).
Клинические проявленияСВ достаточно разнообразны и зависят от локализации сосудистого поражения, фазы заболевания и степени функциональных нарушений органов. Обычно васкулиты имеют острое или подострое начало с первоначальными общими признаками воспалительного процесса и последующим развитием органной патологии. Ранняя диагностика СВ важна для своевременного начала лечения, запоздалая постановка диагноза сопряжена с высоким риском развития необратимых процессов в различных органах и системах. Необходимо подчеркнуть одну особенность, присущую СВ, – это отсутствие специфических лабораторных тестов; в диагностике васкулитов большое значение играет биопсия органов и тканей, в качестве материала для морфологического исследования могут служить биоптаты кожно-мышечного лоскута, легкого, почек, слизистых оболочек верхних дыхательных путей и желудочно-кишечного тракта. Гистологические исследования позволяют уточнить тип поражения сосудов, характер клеточного инфильтрата, локализацию поражения и др.
Для 10 наиболее часто встречающихся форм СВ существуют классификационные критерии, разработанные Американской коллегией ревматологов в 1990 году. Хотя эти критерии и не включают полный спектр проявлений этих заболеваний, тем не менее дают возможность привлечь внимание врача к характерным сочетаниям симптомов (приводятся далее при рассмотрении частных форм васкулитов).
В настоящее время в качестве основных групп препаратов в лечении СВ используются ГКС и цитостатики, позволяющие приостановить или замедлить процесс развития иммунного воспаления. Возможные механизмы противовоспалительного и иммуномодулирующего действия ГКС при васкулитах (Е.Л. Насонов, 1999):
подавление Th1-типа иммунного ответа;
подавление поступления лейкоцитов в зону воспаления за счет снижения экспрессии молекул адгезии на эндотелиальных клетках;
ингибиция транскрипции генов провоспалительных цитокинов (ИЛ-1, 8, ФНО-α);
изменение функциональной активности лейкоцитов и эндотелия;
подавление активности фосфолипазы А2 и ЦОГ-2;
подавление активации, пролиферации и синтеза антител В-лимфоцитами;
стимуляция апоптоза Т- и В-лимфоцитов;
подавление индуцированного ИЛ-1 хемотаксиса нейтрофилов и выработки ими супероксидных радикалов, простагландинов и лейкотриенов.
Другим направлением терапии васкулитов является коррекция микроциркуляторных нарушений, ведущих к вторичным ишемическим изменениям в органах и тканях. При ряде форм СВ, ассоциированных с иммунными комплексами, показаны экстракорпоральные методы очищения крови (плазмаферез).
По мнению ведущих ревматологов, в лечении СВ можно выделить несколько основных этапов:
Быстрое подавление иммунного ответа в начале заболевания – индукция ремиссии – до развития необратимых ишемических, некротических изменений в органах и тканях.
Длительная (не менее 0,5-2 лет) поддерживающая терапия иммуносупрессантами в дозах, достаточных для достижения клинической и лабораторной ремиссии. Быстрое купирование иммунного ответа при обострениях заболевания.
Достижение стойкой, полной ремиссии васкулита, определение степени повреждения органов или систем организма с целью их коррекции, проведение реабилитационных мероприятий.
Монотерапия ГКС – основной метод лечения гигантоклеточного височного артериита и артериита Такаясу, а также системных некротизирующих васкулитов с ограниченным поражением почек и при отсутствии признаков прогрессирования (например, синдром Чарга–Стросса) и криоглобулинемического васкулита. Как правило, в начале заболевания ГКС назначают внутрь в несколько приемов (преимущественно в первой половине дня с учетом циркадного ритма эндогенных ГКС) в дозе 0,75-1-2 мг/кг/сут в пересчете на преднизолон (обычно 60-80 мг/сут). Продолжительность терапии ГКС в такой дозе определяется темпами снижения клинико-лабораторных (и иммунологических) показателей активности болезни: при недостаточной эффективности дозу препарата можно увеличить до 100-120 мг/сут, и только после достижения эффекта (в среднем через 3-4 недели) дозу ГКС начинают уменьшать по 5 мг в 2 недели (с постепенным замедлением скорости снижения) до поддерживающей дозы (из расчета 0,15-0,20 мг/кг/сут, или 10-15 мг/сут), которая назначается на срок от одного года до трех – пяти лет.
У больных, рефрактерных к стандартной терапии, используют пульс-терапию метилпреднизолоном (по 1000 мг внутривенно в течение трех дней подряд), во время пульс-терапии и после нее продолжают прием ГКС внутрь в обычной дозировке. В последнее время в качестве альтернативного средства выбора при проведении пульс-терапии может быть использован дексаметазон, который в 7 раз активнее, чем преднизолон. Кроме того, препарат оказывает выраженное противовоспалительное действие, проявляет минимальную минералокортикоидную активность (не задерживает ионы натрия в организме и слабо влияет на выделение ионов калия). Период его полувыведения составляет 36-54 ч, в то время как для метилпреднизолона он колеблется от 12 до 36 ч. В табл. 33 приведены показания к проведению пульс-терапии ГКС (обычно в сочетании с циклофосфаном) при частных формах СВ.
Таблица 33
Показания к проведению пульс-терапии глюкокортикоидами
при некоторых формах системных васкулитов
(Е.Л. Насонов и соавт., 1999)
|
Заболевания |
Показания |
|
Узелковый полиартериит |
Периферическая гангрена, полинейропатия, поражение ЖКТ, обострения заболевания |
|
Микроскопический полиангиит |
Быстропрогрессирующий гломерулонефрит, обострения заболевания |
|
Гранулематоз Вегенера |
Поражение почек, легких, обострения заболевания |
|
Синдром Чарга–Стросса |
Обострения заболевания |
|
Артериит Такаясу |
Период индукции ремиссии, обострения заболевания, предоперационная подготовка в активную фазу |
|
Геморрагический васкулит |
Поражение ЖКТ, почек, обострения заболевания |
|
Гигантоклеточный артериит Болезнь Бехчета |
Развитие офтальмологических осложнений |
|
Ревматоидный артрит |
Кожный васкулит, дистальная гангрена конечности, полинейропатия, дигитальный артериит |
Препаратами второго ряда в лечении СВ являются цитостатические средства (цитостатики), из них в первую очередь используются циклофосфамид (циклофосфан), азатиоприн (имуран), метотрексат, лейкеран (хлорбутин) и некоторые другие. Цитостатические препараты в отличие от других лекарственных средств вызывают необратимое повреждение клеток. Они элиминируют сенсибилизированные и несенсибилизированные лимфоидные клетки, а также подавляют их функциональную активность (A.S.Faucietal., 1993). Основные механизмы действия циклофосфана при васкулитах (Е.Л. Насонов и соавт., 1999):
вызывает абсолютную Т- и В-лимфопению с преимущественной элиминацией В-лимфоцитов;
подавляет синтез антител В-лимфоцитами;
подавляет активность нейтрофилов, естественных киллеров (NK-клетки), активированныхCD8+ Т-лимфоцитов;
уменьшает экспрессию на поверхности эндотелиальных клеток молекул адгезии.
Лечение циклофосфаном обычно начинают с дозы 1-2 мг/кг/сут (перорально) в течение 10-14 дней с последующим титрованием в зависимости от уровня лейкоцитов в периферической крови (не ниже 3,0-3,5×109/л, для нейтрофилов – содержание в крови не ниже 1,0-1,5×109/л). При проведении цитостатической терапии необходимо иметь в виду, что улучшение состояния может наблюдаться лишь через 3-4 недели с момента начала лечения, а отчетливый клинический эффект – только через 2-3 месяца. При снижении активности заболевания переходят на прием препарата в поддерживающей дозе (по 50-100 мг/сут перорально или 200-400 мг/нед. внутримышечно). Иммуносупрессоры чаще используются не в качестве монотерапии, а в сочетании с ГКС, что позволяет уменьшить дозу последних для предупреждения развития побочных эффектов.
Комбинированное лечение ГКС и циклофосфаном показано пациентам с системными некротизирующими васкулитами (гранулематоз Вегенера, микроскопический полиангиит), а также больным с тяжелыми формами геморрагического васкулита и синдрома Чарга–Стросса в случае наличия быстропрогрессирующего поражения сосудов и почек, даже несмотря на хороший начальный клинический эффект на ГКС.
Доказана эффективность низких доз метотрексата (0,15-0,30 мг/кг/нед.) в сочетании с высокими дозами преднизолона (1 мг/кг/сут) у пациентов с кожной формой узелкового полиартериита и гранулематозом Вегенера.
В табл. 34 представлена общая схема лечения больных тяжелыми формами системных некротизирующих васкулитов.
Таблица 34
Схема лечения системных васкулитов
(Е.Л. Насонов и соавт., 1999)
|
Индукционная терапия(4-6 месяцев) | ||||
|
Циклофосфан 2 мг/кг/сут в течение 1 месяца (максимальная доза 150 мг/сут). Снизить дозу на 25 мг, если пациент старше 60 лет. Количество лейкоцитов должно быть не менее 4,0×109/л |
и |
преднизолон 1 мг/кг/сут (максимальная доза 80 мг/сут); снижать каждую неделю до 10 мг/сут в течение 6 месяцев | ||
|
Поддерживающая терапия | ||||
|
Азатиоприн 2 мг/кг/сут |
и |
преднизолон 5-10 мг/сут | ||
|
Эскалационная терапия* | ||||
|
Плазмаферез 7-10 процедур в течение 14 суток (замена плазмы в объеме 60 мл/кг 4,5-5 % человеческим альбумином) |
или пульс-терапия метилпреднизолоном 1000 мг/сут в течение 3 суток |
или циклофосфан 2,5 мг/кг/сут пациентам не старше 60 лет | ||
|
Примечание: * – активное тяжелое заболевание с повышением креатинина более 500 мкмоль/л или с легочными геморрагиями. | ||||
В качестве еще одного потенциального патогенетического средства в терапии СВ (в частности, гранулематоза Вегенера и микроскопического полиангиита) может выступать иммуноглобулин, вводимый внутривенно в дозе от 0,4 до 2,0 мг/кг/сут в течение 3-5 дней, при необходимости его инфузии повторяют 1 раз каждые 4 недели.
Нарушение микроциркуляции и сопряженные с ним такие процессы, как гиперкоагуляция и вазоконстрикция, – одни из важнейших факторов патогенеза СВ. Возникновение данных нарушений не только определяет тяжесть заболевания, но и создает предпосылки для хронизации процесса, приводя к тяжелым дистрофическим изменениям в органах и тканях. С целью коррекции микроциркуляторных нарушений с учетом ведущего механизма, их вызвавшего, могут быть использованы следующие группы препаратов:
антикоагулянты (прямые и непрямые) – гепарин, низкомолекулярные гепарины, фенилин, варфарин;
антиагреганты – трентал (пентоксифиллин, агапурин), дипиридамол (курантил);
группа НПВП;
периферические вазодилататоры – нитраты, папаверин;
ингибиторы АПФ – каптоприл, эналаприл, лизиноприл, моноприл, берлиприл, фозиноприл, периндоприл и др.
Патогенетическое обоснование применения лекарственных препаратов при СВ приведено на рис. 17.
Р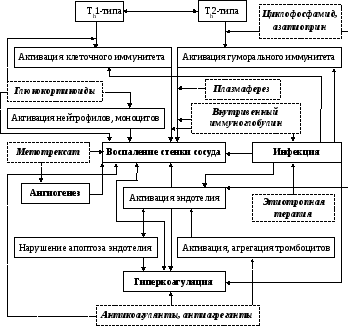 ис.
17. Основные механизмы действия
лекарственных препаратов
ис.
17. Основные механизмы действия
лекарственных препаратов
при васкулитах (цит. по Е.Л. Насонову и соавт., 1999)
Далее будут рассмотрены некоторые частные формы СВ, встречающиеся в практике врача-клинициста.
Узелковый полиартериит(УП) – системный некротизирующий васкулит – заболевание артерий среднего и мелкого калибра с образованием аневризм и вторичным поражением органов и систем. В настоящее время считается, что прежнее название заболевания «узелковый периартериит» не совсем точно отражает сущность процесса, поскольку при нем поражаются все слои сосудистой стенки, а не только адвентиция или окружающие ткани. В связи с этим он заменен на «узелковый полиартериит». Термин «узелковый» отражает образование узелков по ходу пораженного сосуда, т. е. аневризм, являющихся патогномоничным признаком этого заболевания.
Из этиологических факторов важнейшими являются лекарственная непереносимость и персистирование вируса гепатита В. Характерный патоморфологический признак УП, по определению J.T.Lie(1990), – сегментарный некротизирующий васкулит артерий мышечного типа среднего и мелкого калибра, значительно реже артериол и крайне редко венул. В патологический процесс могут быть вовлечены практически любые артерии, однако поражение аорты и других крупных эластических артерий в литературе не описано. В острую фазу заболевания имеет место воспаление стенки артерий с фибриноидным некрозом ее средней оболочки, воспалительный инфильтрат полиморфный (преимущественно представлен нейтрофилами, в меньшей степени – эозинофилами и лимфоцитами). В дальнейшем отмечается полное нарушение нормальной структуры сосудистой стенки, нередко на месте воспалительной инфильтрации формируются аневризмы артерий с пристеночным тромбозом. Участки абсолютно нормальной артерии могут быть расположены рядом с поврежденным сегментом сосуда. Заживление очага поражения сопровождается развитием склеротических изменений в стенке сосуда и окклюзии его просвета.
Заболевание начинается постепенно, реже остро (после приема некоторых лекарственных препаратов), с лихорадки, не купирующейся приемом антибиотиков, миалгии, болей в суставах, кожных высыпаний, похудания, вплоть до прогрессирующей кахексии, что может свидетельствовать о высокой активности болезни. Типична интенсивная боль в икроножных мышцах, иногда приводящая к обездвижению пациента, часто миалгия предшествует появлению нейропатии. Суставной синдром встречается у каждого второго больного, типично поражение крупных суставов по типу транзиторного, недеформирующего артрита, иногда артрит при УП может напоминать таковой при РА. Поражение кожи наблюдается у 25-60 % больных, преобладают сосудистая папулопетехиальная пурпура, сетчатое ливедо, реже встречаются буллезные и везикулезные высыпания.
Самым частым (60-80 %) и прогностически неблагоприятным синдромом при УП является поражение почек (появляется в среднем через 3-6 месяцев от начала заболевания). По современным представлениям, при классическом УП преобладает сосудистый тип почечной патологии, тогда как развитие гломерулонефрита наблюдается значительно чаще при микроскопическом полиангиите. Воспалительные изменения затрагивают междолевые артерии и реже артериолы. Быстрое формирование и неуклонное прогрессирование ХПН, как правило, связано с множественными (безболевыми) инфарктами почек. Наиболее частыми признаками поражения почек являются умеренная протеинурия (не превышающая за сутки 3 г), микрогематурия (как показатель активности болезни), лейкоцитурия (не связанная с мочевой инфекцией). Примерно в 70 % случаев нефропатия при УП сопровождается артериальной гипертензией, развитие которой на ранних стадиях обусловлено васкулитом или инфарктом почки, тогда как на поздних – вторичным поражением клубочков. Артериальная гипертензия при УП стойкая и не исчезает в период ремиссии заболевания, носит ангиотензинзависимый характер (эффективны препараты, ингибирующие АПФ).
Абдоминальный синдром – второй диагностически значимый и прогностически серьезный признак, свидетельствующий о генерализации болезни. Он обусловлен сосудистыми расстройствами и ишемией органов брюшной полости, встречается у 36-44 % больных. В клинической картине доминируют такие симптомы, как тошнота, рвота, постоянная интенсивная боль, диарея, анорексия; в 10-13 % случаев при абдоминальном синдроме развиваются инфаркты брыжейки или кишки, язвы, часто с перфорацией, перитонит, реже (в 6-7 %) – желудочно-кишечные кровотечения. Поражение печени проявляется ее увеличением и изменением содержания печеночных ферментов в крови, все это может быть связано как с инфекцией вирусом гепатита В, так и с инфарктом печени или гематомой в результате разрыва внутрипеченочных сосудов.
Третьим классическим признаком УП является поражение периферической нервной системы, встречающееся в 50-60 % случаев. Уже в первые месяцы заболевания возникают периферические невриты, связанные с поражением vasa vasorumв результате васкулита. Чаще поражаются малоберцовый нерв, лучевой, реже локтевой и срединный. Для УП характерны множественные асимметричные невриты, что проявляется выраженной атрофией мышц конечностей, парезами кистей и стоп, резким снижением сухожильных рефлексов.
При УП возможно вовлечение в патологический процесс других органов и систем, таких как сердце, легкие, ЦНС, но встречаются эти поражения намного реже, чем описанная выше триада клинических симптомов.
Не существует каких-либо специфических лабораторных тестов для диагностики УП. Как уже было сказано ранее, лабораторные изменения в основном отражают активность болезни (повышение острофазовых белков, лейкоцитоз, тромбоцитоз) и степень сосудистых повреждений в отдельных органах. Огромное значение уделяется морфологической диагностике этого васкулита, однако необходимо помнить о том, что поражение сосудов при УП носит сегментарный характер, вследствие чего следует проводить биопсию большого количества органов и тканей (кожи, слизистой кишечника, мышц, почек, печени).
Критерии диагностики классического УП представлены в табл. 35.
Таблица 35
Классификационные критерии УП (R.Lightfootetal., 1990)
|
Критерий |
Определение |
|
Потеря 4 кг и более со времени начала заболевания, не связанная с диетой и другими факторами |
|
Наличие сетчатых участков на коже конечностей и туловища |
|
Боль или болезненность в тестикулах, не связанные с инфекцией, травмой или другими причинами |
Окончание табл. 35
|
Критерий |
Определение |
|
Диффузные миалгии (включая плечевой и тазовый пояса), мышечная слабость, болезненность мышц ног |
|
Развитие моно- или полинейропатии |
|
Развитие артериальной гипертензии с диастолическим давлением выше 90 мм рт.ст. |
|
Повышение азота мочевины >40 мг/дл или креатинина >1,5 мг/дл, не связанное с дегидратацией или обструкцией почечных сосудов |
|
Обнаружение антител к вирусу гепатита В в сыворотке крови или антигена вируса гепатита В |
|
Наличие аневризм или окклюзий висцеральных артерий, не связанное с артериосклерозом, фибромышечной дисплазией или другими неинфекционными причинами |
|
Гистологические изменения с наличием гранулоцитов или мононуклеаров в стенке сосуда |
Наличие у больного 3 и более любых критериев позволяет поставить диагноз УП с чувствительностью 82,2 % и специфичностью 86,6 %. Добавление в качестве 11-го критерия абдоминального болевого синдрома или перфорации кишки увеличивает чувствительность критериев до 83,9 %, но снижает специфичность до 84,9 %.
Правильная формулировка диагноза сводится к точному нозологическому определению болезни, варианта течения (медленно прогрессирующее или быстропрогрессирующее), степени активности и развившихся осложнений.
Пятилетняя выживаемость больных УП при проведении активной иммуносупрессивной терапии составляет в среднем 60-90 %, у нелеченных – лишь до 13 %. Причинами смерти чаще всего являются: хроническая почечная недостаточность (каждый второй пациент), церебральные расстройства (10-12 %), сердечная недостаточность (13-15 %), поражение ЖКТ (12-15 %).
Ведение больных УП, как и другими системными некротизирующими васкулитами, зависит от тяжести и распространенности сосудистой патологии. При ограниченном поражении сосудов в отсутствие клинических признаков быстрого прогрессирования заболевания назначаются средние дозы ГКС, тогда как у больных с быстропрогрессирующим вариантом течения целесообразно раннее назначение комбинированной терапии ГКС и цитостатиками (предпочтение отдают циклофосфамиду).
В случае носительства антигена вируса гепатита В ведение больных УП в целом не отличается от такового у других пациентов, однако при обнаружении серологических маркеров активной репликации вируса гепатита В показано назначение противовирусных препаратов (интерферон-α, видарабин, ламивудин) в сочетании со средними дозами ГКС и повторными курсами плазмафереза, тогда как применение высоких доз цитостатиков менее целесообразно (Е.Н. Семенкова, 1988; Е.Л. Насонов, 1999).
Микроскопический полиангиит(МПА) – это некротизирующий васкулит с небольшим количеством или отсутствием иммунных депозитов, поражающий преимущественно мелкие сосуды (артериолы, капилляры, венулы), в клинической картине которого доминируют явления некротизирующего гломерулонефрита и легочные капилляриты (J.C.Jennetteetal., 1994). Эта форма СВ в силу особенностей клинической картины, морфологических и иммунных нару-шений выделена из УП. МПА встречается в 10 раз чаще, чем классический УП, и в 2 раза чаще, чем гранулематоз Вегенера.
В патогенезе этого васкулита ведущая роль отводится образованию АНЦА, обладающих антиэндотелиальной активностью. Необходимо отметить, что в отличие от УП, некротизирующий васкулит при МПА носит генерализованный характер, при этом наиболее выраженные изменения наблюдаются в сосудах микроциркуляции кожи, легких и почек.
В дебюте заболевания могут появляться такие же неспецифические симптомы, как и при УП, однако уже на ранних этапах болезни в процесс вовлекаются почки. Их поражение характеризуется фокальным сегментарным некротизирующим экстракапиллярным гломерулонефритом с «полулуниями», часто приобретающим быстропрогрессирующее течение с исходом в ХПН. Особенности поражения почек при этих двух формах СВ представлены в табл. 36.
Таблица 36
Особенности поражения почек при классическом УП и МПА
|
Признак |
УП |
МПА |
|
Мочевой синдром |
60 – 80 % |
В 100 % с последующей трансформацией в нефротический синдром |
|
Реноваскулярная гипертензия |
Часто |
Редко |
|
Злокачественная АГ |
10 % |
Не бывает |
|
Инфаркты почек |
Встречаются |
Не бывает |
|
Микроаневризмы |
Часто |
Не бывает |
|
Быстропрогрессирующий гломерулонефрит |
Очень редко |
Часто |
|
Хроническая почечная недостаточность |
Развивается на поздних стадиях |
Быстрое развитие на ранних этапах болезни |
|
Ангиография почечных сосудов (микроаневризмы, стенозы) |
Часто (до 60 % случаев) |
Не бывает |
Вторым по частоте (30-50 %) клиническим признаком является поражение легких. Больных беспокоят одышка смешанного характера, кровохарканье, боли в грудной клетке при дыхании. На рентгенограммах органов грудной клетки могут обнаруживаться легочные инфильтраты с реакцией плевры без признаков распада (при динамическом наблюдении), быстро исчезающие при использовании системных ГКС.
До сих пор не существует классификационных критериев МПА, и его диагноз основывается на совокупности клинических, морфологических и иммунологических данных. Исходя из знаний патогенеза этого заболевания, специфическим иммунологическим маркером МПА считаются перинуклеарные АНЦА, направленные против миелопероксидазы, которые обнаруживаются почти в 80 % случаев.
В лечении используются ГКС в сочетании с цитостатиками в общепринятых дозировках. В случае быстрого и неуклонного прогрессирования ХПН показано проведение программного гемодиализа. Прогноз при МПА несколько хуже, чем при УП, пятилетняя выживаемость составляет в среднем 65 %, несмотря на адекватную комбинированную терапию ГКС и иммунодепрессантами.
Синдром Чарга–Стросса– эозинофильное гранулематозное воспаление респираторного тракта и некротизирующий васкулит, поражающий мелкие и средние сосуды, часто сочетающийся с астмой и эозинофилией (J.C.Jennetteetal., 1994). В последние годы на основании клинических, патологических особенностей и иммунологических нарушений выделен в отдельную нозологическую форму из УП.
Патогенез связан с развитием реакций гиперчувствительности немедленного типа, в которой основная роль принадлежит иммуноглобулинам класса Е; адсорбируясь на поверхности тучных клеток и базофилов, они вызывают их дегрануляцию и выделение в кровь биологически активных веществ. Роль эозинофилов заключается в активном подавлении медиаторов тучных клеток и фагоцитозе их гранул, однако в гранулах самих эозинофилов содержатся вещества, способные повреждать слизистые оболочки, эндотелий сосудов и эндокард.
В течение заболевания условно можно выделить 3 основных фазы (J.Lachman, 1984). В продромальный период, который может длиться до 30 лет, у больных имеют место различные аллергические реакции, включающие ринит, поллиноз и астму. Второй период характеризуется эозинофилией крови и тканей, в третьей фазе в клинической картине болезни превалируют признаки СВ.
Основное проявление заболевания – синдром гиперреактивности бронхов, который в большинстве случаев предшествует развитию развернутой клинической картины, обусловленной СВ. На ранних этапах заболевания бронхоспазм выражен умеренно, в дальнейшем наблюдается развитие тяжелой бронхиальной астмы, более чем в 70 % случаях требующей назначения системной терапии ГКС. Кроме того, поражение легких характеризуется транзиторными (реже персистирующими) легочными инфильтратами (у 2/3 больных) или эозинофильным экссудатом в плевральной полости (у 1/3 пациентов).
Поражение сердца встречается достаточно часто (47-56 %), нередко в виде развития эндокардиального фиброза, приводящего к появлению и прогрессированию сердечной недостаточности и тромбоэмболических осложнений.
Патология нервной системы в основном не отличается от таковой при УП и включает как поражения периферических нервов (в виде множественных мононевритов или симметричной сенсорно-моторной периферической полинейропатии), так и изменения со стороны черепно-мозговых нервов (особенно часто наблюдается ишемия зрительного нерва).
Вовлечение в процесс ЖКТ проявляется болями в животе и диареей, иногда кровотечением, все эти синдромы обусловлены как эозинофильным гастроэнтеритом, так и васкулитом стенки кишки, причем последний может привести к перфорации кишечника, перитониту и кишечной непроходимости.
Поражение почек встречается реже и протекает менее злокачественно, чем при гранулематозе Вегенера или МПА. У половины больных выявляется очаговый нефрит, часто приводящий к повышению АД. У пациентов с положительными серологическими АНЦА отмечается развитие некротизирующего гломерулонефрита.
Изменения кожи относятся к одному из наиболее характерных проявлений заболевания. При синдроме Чарга–Стросса они встречаются даже чаще, чем при классическом УП. К ним относятся узелки, пурпура, эритема, крапивница, кожные некрозы и сетчатое ливедо. У половины больных наблюдается поражение суставов в виде полиартрита или полиартралгий. Характерен непрогрессирующий мигрирующий артрит крупных и мелких суставов. Изредка встречаются миалгии и миозит.
Лабораторным маркером заболевания является эозинофилия (более 1×109/л), которая встречается у 97 % больных на любой стадии заболевания. Отмечается взаимосвязь между уровнем эозинофилов в периферической крови и выраженностью клинических проявлений астмы и васкулита. Закономерным также является повышение концентрации сывороточногоIgE. В миелограмме повышено количество зрелых эозинофилов.
Классификационные критерии синдрома Чарга–Стросса приведены в табл. 37.
Наличие у больного любых 4 и более критериев позволяет поставить диагноз с чувствительностью 85 % и специфичностью 99 %.
Таблица 37
Классификационные критерии синдрома Чарга–Стросса (A.T.Masietal., 1990)
|
Критерий |
Определение |
|
Затруднение дыхания или диффузные хрипы на выдохе |
|
Эозинофилия более 10 % при подсчете лейкоцитарной формулы |
|
Сезонная аллергия (аллергический ринит) или другие аллергические реакции (пищевая, контактная), за исключением лекарственной |
|
Мононейропатия, множественная полинейропатия по типу перчаток (чулок) |
|
Мигрирующие или транзиторные легочные инфильтраты, обнаруженные при рентгенологическом обследовании |
|
Боли в области придаточных пазух носа или рентгенологические изменения |
|
Скопление эозинофилов во внесосудистом пространстве (по данным биопсии) |
Заболевание может быть заподозрено у пациентов среднего возраста с длительно текущей бронхиальной астмой, аллергическим ринитом и эозинофилией при развитии у них признаков системной патологии, включающей полиневрит, легочные инфильтраты, кардиомиопатию.
Дифференциальный диагноз заболевания следует проводить с УП, гранулематозом Вегенера, хронической эозинофильной пневмонией. В отличие от синдрома Чарга–Стросса, при классическом УП обычно не наблюдается поражения легких, астмы и некротизирующего гломерулонефрита. Дифференциальный диагноз синдрома Чарга–Стросса и гранулематоза Вегенера также не представляет трудностей, для последнего не характерно развитие астмы, аллергических реакций и эозинофилии. Поражение ЛОР-органов при синдроме Чарга–Стросса встречается достаточно часто, но не сопровождается некротическими изменениями.
Лечение синдрома Чарга–Стросса основывается на тех же принципах, что и УП. В зависимости от тяжести заболевания преднизолон назначается в дозе 40-60 мг/сут в течение нескольких недель с постепенным снижением дозы. При недостаточной эффективности ГКС используют циклофосфан, азатиоприн или хлорбутин в общепринятых дозировках. Прогноз нелеченных больных неблагоприятный: только 20 % пациентов живут более 2 лет, применение цитостатической терапии позволило добиться 10-летней выживаемости у 75 % больных синдромом Чарга–Стросса.
Гранулематоз Вегенера– гранулематозное воспаление респираторного тракта и некротизирующий васкулит, поражающий мелкие и средние сосуды (капилляры, венулы, артериолы, артерии), обычно сочетающийся с некротизирующим гломерулонефритом (J.C.Jennetteetal., 1994).
Гранулематозом Вегенера одинаково часто болеют как мужчины, так и женщины. Средний возраст заболевших обычно составляет около 40 лет. В дебюте и развернутой стадии заболевания, как правило, наблюдаются общевоспалительные синдромы: лихорадка, общая слабость, похудение, миалгии, артралгии и реже артриты. Для гранулематоза Вегенера характерна классическая триада – поражение верхних дыхательных путей, легких, почек.
Поражение ЛОР-органов является самым частым (70 %) и нередко первым признаком болезни. Возможны – стойкий насморк с гнойно-кровянистыми выделениями из носа, заложенность носа, сухие корки, носовые кровотечения, потеря обоняния, в дальнейшем при прогрессировании процесса развиваются язвенно-некротические изменения слизистых оболочек с вовлечением глотки, гортани, трахеи, а также гайморовых пазух с разрушением хряща и костной ткани носовой перегородки, верхнечелюстной пазухи и орбиты.
Изменения в легких встречается у 85 % больных, причем у 45 % – в дебюте заболевания. Они характеризуются множественными двусторонними инфильтратами, склонными к распаду и формированию полостей. Реже наблюдаются плеврит, легочные кровотечения и увеличение лимфатических узлов средостения. Только в 65 % случаев рентгенологические признаки патологии легких сочетаются с клиническими (кашель, кровохарканье, одышка), у остальных же больных они протекают без субъективных проявлений.
Поражение почек – третий классический признак гранулематоза Вегенера, и если в дебюте заболевания гломерулонефрит наблюдается лишь у 11-18 %, то в развернутую стадию – у 77-85 % больных. Такие симптомы поражения почек, как протеинурия, гематурия и другие, появляются через несколько месяцев от начала болезни, что подтверждает иммунный механизм их повреждения. Необходимо подчеркнуть, что, несмотря на адекватную цитостатическую терапию, более чем у 40 % больных гранулематозом Вегенера развивается ХПН, требующая проведения программного гемодиализа или пересадки почек.
Среди патологии других органов существенное клиническое и диагностическое значение имеют поражения глаз (односторонний экзофтальм, склерит, увеит, окклюзия центральной артерии сетчатой оболочки), органа слуха (средний отит, гранулематозное разрушение височных костей), периферической (полиневрит, дистальная симметричная полинейропатия) и ЦНС, включая II,V,VII,VIII,IXиXIIпары черепных нервов и мозговые оболочки.
Классификационные критерии гранулематоза Вегенера представлены в табл. 38.
Таблица 38
Классификационные критерии гранулематоза Вегенера (R.Y.Leavittetal., 1990)
|
Критерий |
Определение |
|
Развитие болезненных или безболезненных язв в ротовой полости, гнойные или геморрагические выделения из носа |
|
Наличие узелков, инфильтратов или каверн |
|
Микрогематурия (5 эритроцитов в поле зрения или их скопления) |
|
Гранулематозное воспаление в стенке артерий или артериолах, или в периваскулярных и экстраваскулярных зонах |
Наличие 2 из 4 критериев свидетельствует о достоверном гранулематозе Вегенера. Чувствительность любых двух и более критериев составляет 88,2 %, а специфичность – 92,0 %.
Лабораторные изменения при гранулематозе Вегенера, как и при других СВ, неспецифичны. В период активности заболевания наблюдаются нормохромная анемия, тромбоцитоз, нейтрофильный лейкоцитоз, повышение СОЭ, гипергаммаглобулинемия, положительные СРБ и РФ. Среди иммунологических показателей определяются повышенные уровни сывороточных иммуноглобулинов, особенно IgA, циркулирующих иммунных комплексов и АНЦА.
При гранулематозе Вегенера особое значение принадлежит мор-фологическому подтверждению диагноза, в связи с чем необходимо проводить биопсию пораженных органов, при этом диагностическая значимость открытой биопсии легкого гораздо выше трансбронхиальной или морфологического исследования тканей слизистой оболочки носа и его придаточных пазух. При отсутствии результатов гистологического исследования или неинформативности полученных данных в критерии заболевания вводится дополнительный клинический признак – кровохарканье.
Стандартная терапия гранулематоза Вегенера, предложенная A.S.Fauciи соавт. еще в 1983 г., основана на пероральном приеме циклофосфана (из расчета 2-3 мг/кг/сут) в сочетании с преднизолоном (1 мг/кг/сут). Лечение ГКС в этой дозе продолжают до развития иммуносупрессивного эффекта цитостатика (в среднем около 4 недель), затем дозу преднизолона постепенно снижают по 5 мг каждые 2 недели до поддерживающей (5-10 мг/сут), которая сохраняется и при достижении ремиссии заболевания. Несмотря на достигнутый полный контроль над течением васкулита, лечение циклофосфаном продолжают на протяжении, по крайней мере, еще 1 года, после чего дозу препарата уменьшают постепенно на 25 мг каждые 2-3 месяца. Ввиду высокого риска развития побочных эффектов от приема циклофосфана (геморрагический цистит, рак мочевого пузыря) в настоящее время широко используют другие иммунодепрессанты, такие как азатиоприн, метотрексат, лейкеран. Была показана определенная эффективность циклоспорина А в начальной дозе 5 мг/кг/сут (в сочетании с низкими дозами ГКС) у больных гранулематозом Вегенера, у которых предшествующее лечение циклофосфаном было недостаточным для индукции ремиссии или ассоциировалось с тяжелыми побочными эффектами (N.Allenetal., 1993). Однако в этом же исследовании было отмечено, что снижение дозы циклоспорина А до поддерживающей (1-2 мг/кг/сут) сопровождалось обострением заболевания у данных пациентов.
В отсутствие лечения средняя продолжительность жизни больных гранулематозом Вегенера составляет лишь 5 месяцев, а смертность среди них в течение первого года заболевания достигает 80 %. Рано начатое лечение позволяет существенно улучшить прогноз болезни: пятилетняя выживаемость при монотерапии ГКС составляет 12 %, а при комбинации ее с цитостатиками – 61 % (по данным Е.Н. Семенковой, 1988). Тем не менее смертность больных гранулематозом Вегенера продолжает оставаться на высоком уровне, наиболее частыми причинами летальных исходов являются интеркуррентные инфекции, дыхательная и почечная недостаточность.
Синдром Гудпасчера– системный васкулит с преимущественным поражением легких и почек по типу пневмонита и гломерулонефрита. Данная форма васкулита чаще встречается у молодых мужчин, соотношение между мужчинами и женщинами составляет от 5:1 до 9:1.
Ряд авторов признает связь васкулита с вирусной (вирусы гриппа) или бактериальной (стрептококки типа А12) инфекциями. Заболевание имеет аутоиммунный патогенез: в качестве антигенов выступают базальные мембраны собственных альвеол и почечных клубочков, против которых вырабатываются аутоантитела.
Начало заболевания, как правило, острое и проявляется легочной патологией (в 75 % случаев): рецидивирующим кровохарканьем (вплоть до развития легочного кровотечения), кашлем и одышкой на фоне неспецифических симптомов, таких как лихорадка, потливость, общая слабость, выраженная утомляемость. Вслед за легочной патологией развивается поражение почек (реже оно является первым признаком заболевания), характеризующееся гематурией, протеинурией, олигоурией и быстрым формированием почечной недостаточности. Выраженная артериальная гипертензия не свойственна. Еще одним характерным клиническим признаком этой формы васкулита является гипохромная анемия, причинами которой могут быть рецидивирующие кровохарканья, геморрагии в легочные альвеолы, уменьшение выработки эритропоэтина почками на фоне ХПН.
Диагностика синдрома Гудпасчера основывается на клинических данных (преимущественное поражение легких и почек) и обнаружении антител к базальным мембранам альвеол и клубочков почек. В лечении актуально назначение высоких доз преднизолона (из расчета 1 мг/кг массы тела) в сочетании с циклофосфаном или азатиоприном в дозе 150-200 мг/сут.
Неспецифический аортоартериит(или болезнь Такаясу, по имени японского офтальмолога, впервые описавшего это заболевание у молодой женщины, сопровождающееся изменениями центральной артерии сетчатой оболочки и отсутствием пульса на плечевой артерии) – гранулематозное воспаление аорты и ее основных ветвей, обычно начинающееся в возрасте до 50 лет (J.C.Jennetteetal., 1994).
Болезнь встречается преимущественно у молодых женщин (в 80-90 % случаев), средний возраст составляет от 10 до 30 лет.
Типичная локализация патологического процесса – аорта и ее главные стволы, включая коронарные и почечные артерии, а также легочные артерии эластического типа, с поражением адвентиции, внутренней части средней оболочки артерий и области vasavasorum. Воспалительный инфильтрат представлен в основном лимфоцитами, плазматическими клетками и гигантскими многоядерными клетками. Процесс завершается склерозированием, дегенерацией медии и фиброзом адвентиции, в 20 % случаев возможно образование аневризмы. Было показано, что по мере прогрессирования артериита формируются сегментарные стенозы и окклюзии сосудов, приводящие к развитию ишемии ниже места сужения сосудов. В связи с медленным развитием болезни и сегментарным типом поражения обычно формируется хорошая коллатеральная сеть.
Клинические проявления артериита Такаясу условно можно подразделить на две группы. В первую входят неспецифические симптомы, обусловленные системным воспалительным ответом организма, наблюдающиеся в дебюте заболевания или во время его обострений (длительная лихорадка, похудение, слабость, сонливость, миалгии и артралгии и др.). Ко второй группе относят синдромы, в основе которых лежат ишемические изменения в отдельных органах или тканях, обусловленные прогрессирующим поражением артерий.
Наиболее часто поражаются ветви дуги аорты (до 80 % всех случаев). Так, при локализации воспалительного процесса в сосудах верхних конечностей отмечается похолодание, онемение и слабость в руках с постепенным нарастанием гипотрофии мышц плечевого пояса. При объективном осмотре определяется ослабление или отсутствие пульса на одной или обеих руках с характерной асимметричностью поражения. В случае окклюзии сонных артерий типичны жалобы на головокружение, головные боли, возможно развитие синкопальных состояний. При сужении нисходящего отдела аорты основным клиническим признаком будет различие уровней систолического АД на верхних и нижних конечностях.
Поражение сердца встречается у каждого второго больного, кардиальные симптомы проявляются инфарктом миокарда, недостаточностью клапанов аорты, сердечной недостаточностью, артериальной гипертензией, причинами которой могут быть стеноз устья почечной артерии или сужение просвета аорты и снижение эластичности ее стенок.
Поражение ЖКТ (10-25 %) обусловлено абдоминальной ишемией и проявляется болью в эпигастральной или мезогастральной области, возникающей через полчаса после приема пищи и купирующейся самостоятельно через 2 часа.
Поражения органа зрения проявляются в виде быстро проходящей слепоты или снижения остроты зрения.
Поражение бифуркации аорты нередко сопровождается вовлечением в процесс подвздошных и бедренных артерий и клинически выражается в ишемии нижних конечностей с явлениями перемежающейся хромоты, отсутствии или резком ослаблении пульсации артерий, понижении АД на ногах, систолическом шуме над пораженными артериями.
K.Ishikawaи соавт. (1981) предложили классифицировать течение артериита Такаясу в зависимости от наличия и степени выраженности четырех основных осложнений, непосредственно связанных с данным заболеванием, к которым относятся ретинопатия, артериальная гипертензия, аортальная недостаточность и аневризмы. Больные подразделяются на 4 основные группы:
1-я группа – пациенты, не имеющие перечисленных выше синдромов (10-летняя выживаемость составляет 97 %);
2-я группа (А) – пациенты, имеющие одно осложнение (легкие или умеренно выраженные осложнения, под ними подразумевают первую стадию ангиопатии сетчатки, артериальную гипертензию менее 200/110 мм рт.ст. на верхних конечностях, диаметр аневризмы размером, не превышающим два просвета сосуда, и аортальную недостаточность 1–2-й степени) (10-летняя выживаемость составляет 97 %);
2-я группа (Б) – пациенты, имеющие одно осложнение (тяжелые осложнения) (10-летняя выживаемость составляет 58,6 %);
3-я группа – пациенты, имеющие два или более основных осложнений (10-летняя выживаемость составляет 58,6 %).
Специфических лабораторных тестов для диагностики неспецифического аортоартериита не существует, большое значение для дифференциального диагноза имеет ангиография, которая должная выполняться у всех пациентов с подозрением на данную форму васкулита. Классификационные критерии заболевания приведены в табл. 39.
Таблица 39
Классификационные критерии болезни Такаясу
(W.P. Arend et al., 1990)
|
Критерий |
Определение |
|
Развитие первых признаков неспецифического аортоартериита у пациентов моложе 40 лет |
|
Развитие или усиление мышечной слабости или дискомфорта в одной или в обеих конечностях при движении |
|
Ослабление пульсации на одной или обеих плечевых артериях |
|
Различия систолического давления на руках более 10 мм рт.ст. |
|
Сосудистый шум при аускультации одной или обеих подключичных артерий или брюшной аорты |
|
Сужение просвета или окклюзия самой аорты или ее основных стволов, или крупных артерий в проксимальной части на верхних или нижних конечностях, не связанные с артериосклерозом; изменения обычно носят сегментарный характер |
При наличии 3 и более критериев из 6 диагноз можно считать достоверным. Чувствительность трех и более критериев составляет 90,5 %, специфичность – 97,8 %.
Лечение артериита Такаясу направлено на подавление острого воспаления в сосудистой стенке, профилактику возможных осложнений и компенсирование симптомов сосудистой недостаточности. В начале болезни обычно назначают преднизолон в дозе 1 мг/кг/сут в течение 1 месяца с последующим постепенным снижением дозы до поддерживающей (10 мг/сут), которую принимают на протяжении 2-5 лет. В лечении неспецифического аортоартериита широко используют антикоагулянты и сосудорасширяющие препараты, однако их эффективность не доказана.
В случаях наличия у больного симптомов расслоения аневризмы брюшной аорты, двусторонних гемодинамически значимых стенозах и/или окклюзиях сонных артерий, выраженной артериальной гипертензии в результате коарктации аорты или при сужении одной почечной артерии показано хирургическое лечение.
Геморрагический васкулит(пурпура Шенлейна–Геноха) – васкулит сIgA-иммунными депозитами, поражающий мелкие сосуды (капилляры, венулы, артериолы). Типичны изменения со стороны кожи, кишечника и почек в сочетании с артралгиями или артритом (J.C.Jennetteetal., 1994).
Геморрагический васкулит, один из наиболее часто встречающихся СВ, относится к группе гиперергических васкулитов, отличительными чертами которых являются обязательное поражение мелких сосудов кожи и связь с аллергией к определенным антигенам. Болезнь может начинаться в любом возрасте, однако преимущественно ею болеют дети до 16 лет. Имеется определенная связь между развитием васкулита и инфекцией верхних дыхательных путей, которая предшествует началу заболевания, по данным Н.П. Шилкиной и соавт. (1990), у 66-80 % больных.
В этиологии геморрагического васкулита обсуждается роль различных микроорганизмов, включая стрептококки, микоплазму, иерсинии, легионеллы, вирусы Эбштейна–Барра и гепатита В, аденовирус, цитомеговирус и парвовирус В19. Другими потенциальными пусковыми агентами являются лекарственные препараты (пенициллин, эритромицин, ампициллин, хинидин и др.), пищевая аллергия, укусы насекомых, действие очень низких или высоких температур. Характерным признаком заболевания является высокий уровень IgAв сыворотке крови. Во многих случаях удается установить наличиеIgA-содержащих иммунных комплексов как в сыворотке крови, так и в очагах кожных и почечных поражений.
Принято выделять следующие основные синдромы болезни Шенлейна–Геноха: кожный, суставной, абдоминальный и почечный. Все перечисленные синдромы могут сочетаться между собой в разной степени выраженности, при этом в случае обострения (или рецидива) заболевание может проявиться качественно новой симптоматикой, в том числе с вовлечением в процесс других органов и систем (легких, сердца, ЦНС).
Поражение кожи – один из диагностических критериев заболевания. Клинические проявления кожного синдрома включают симметричную петехиальную сыпь и/или пурпуру (так называемая пальпируемая нетромбоцитопеническая пурпура) с преимущественной локализацией на разгибательных поверхностях нижних конечностей, ягодицах, нижней половине туловища (никогда не появляются на коже лица). При надавливании элементы сыпи не исчезают, появление новых высыпаний сопровождается неинтенсивным зудом. Обычно через несколько дней пурпура бледнеет, оставляя после себя участки гиперпигментации.
Полиартралгии и артрит встречаются в 59-100 % случаев, чаще у взрослых. Обычно поражение суставов сочетается с миалгиями и отеком голеней. Характерны недлительные (сроком до 1 недели) мигрирующие боли в крупных суставах нижних конечностей, реже локтевых и лучезапястных, возникающие одновременно с геморрагическими высыпаниями.
Поражение желудочно-кишечного тракта наблюдается более чем у 2/3 больных и проявляется спастическими болями в животе, тошнотой, рвотой, реже – желудочно-кишечными кровотечениями. Данные ультразвукового исследования кишечника, полученные A.Coutureи соавт. (1992), свидетельствуют о том, что острые боли в животе связаны с диффузным или очаговым отеком стенки тонкого кишечника. Эндоскопически же обнаруживают геморрагический или эрозивный дуоденит, иногда эрозии в желудке, тонком или толстом кишечнике.
Частота поражения почек при геморрагическом васкулите колеблется от 10 до 60 %. Клинические проявления разнообразны, обычно выявляются изолированная микро- или макрогематурия, сочетающаяся с умеренной протеинурией. В большинстве случаев эти изменения проходят бесследно, но у некоторых больных может развиться гломерулонефрит.
Критерии диагноза геморрагического васкулита представлены в табл. 40.
Таблица 40
Классификационные критерии болезни Шенлейна–Геноха
(J.A. Mills et al., 1990)
|
Критерий |
Определение |
|
Возвышающаяся над поверхностью кожи геморрагическая пурпура, не связанная с тромбоцитопенией |
|
Возраст пациента при появлении первых симптомов болезни – 20 лет и моложе |
|
Диффузные боли в животе, возникающие после еды, обычно сопровождающиеся поносом с кровью |
|
Обнаружение гранулоцитов в стенке артериол и венул |
При наличии более 2 критериев диагноз геморрагического васкулита можно считать достоверным с чувствительностью 87,1 % и специфичностью 87,7 %.
При выборе терапии следует учитывать то, что болезнь Шенлейна–Геноха относится к гиперергическим васкулитам, а следовательно, в некоторых случаях не требует активного лечения. Поражение кожи и суставов обычно хорошо поддается лечению НПВП. Развитие тяжелых системных проявлений заболевания (абдоминальный синдром, гломерулонефрит, кровохарканье и др.) диктует необходимость назначения ГКС.
При всех вариантах геморрагического васкулита проводят лечение гепарином в начальной суточной дозе 300-400 ЕД/кг массы тела под кожу живота через каждые 6 часов под контролем активированного частичного тромбопластинового времени (АЧТВ). Удвоение этого показателя (в норме 35-45 с) свидетельствует об адекватности гепаринотерапии. Часто при высокой активности заболевания начальная доза гепарина оказывается неэффективной, в связи с чем дозу препарата следует постепенно увеличивать по 100 ЕД/кг/сут до общей суточной дозы 800 ЕД./кг массы тела.
Несмотря на достаточно частое рецидивирование заболевания (в 40 % случаев), в целом прогноз при геморрагическом васкулите благоприятный. Пятилетняя выживаемость больных составляет почти 100 %. По данным R.Blancoи соавт. (1997), в течение первых двух лет болезни полное выздоровление наблюдается у 94 % детей и 89 % взрослых. Основным фактором, определяющим неблагоприятный прогноз болезни, является персистирующее поражение почек, которое наблюдается у 2-5 % больных.