

6.4.4. Химические свойства
Общим в химических свойствах карбоновых кислот и их ацильных производных являются реакции в ацильной группе, так как электронное строение функциональной группы кислот и их ацильных производных близкó и различается только величиной электронных эффектов.
Принципиальные различия в строении карбоновых кислот и их ацильных производных связаны с наличием подвижных атомов водорода у электроотрицательного атома (как в случае карбоновых кислот и их амидов) и проявлением ими кислотных свойств.
6.4.4.1. Кислотно-оснόвные свойства
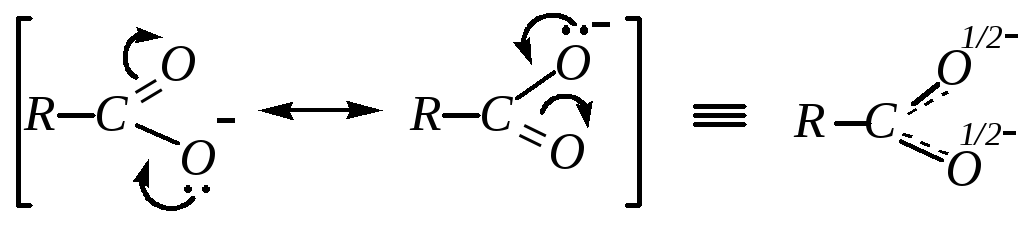
Кислотные свойства карбоновых кислот связаны с наличием сильнополярной связи O–Н. Кроме того, из рассмотрения граничных структур молекулы карбоновой кислоты следует, что возможна миграция протона от одного атома кислорода к другому. А так как природа этих атомов идентична, то состояние реальной молекулы будет описываться некоторой промежуточной формулой, в которой катион водорода в равной мере принадлежит каждому атому кислорода:

При отщеплении от молекулы катиона водорода образуется карбоксилат-анион, стабилизированный p--сопряжением:

С пособствовать
делокализации отрицательного заряда
в этом анионе будут электроноакцепторные
заместители. Поэтому в гомологическом
ряду предельных монокарбоновых кислот
кислотные свойства будут уменьшаться
(из-за возрастания донорного эффекта
радикала R), но
введение в углеводородный радикал
электроноакцепторных заместителей
приведёт к дальнейшей поляризации связиO–Н, более легкому
отрыву протона, стабилизации аниона
кислотного остатка и, как следствие, —
к возрастанию кислотных свойств.
пособствовать
делокализации отрицательного заряда
в этом анионе будут электроноакцепторные
заместители. Поэтому в гомологическом
ряду предельных монокарбоновых кислот
кислотные свойства будут уменьшаться
(из-за возрастания донорного эффекта
радикала R), но
введение в углеводородный радикал
электроноакцепторных заместителей
приведёт к дальнейшей поляризации связиO–Н, более легкому
отрыву протона, стабилизации аниона
кислотного остатка и, как следствие, —
к возрастанию кислотных свойств.
Примером проявления кислотных свойств является образование солей при растворении карбоновых кислот в водных растворах щелочей (известно, что спирты со щелочами не взаимодействуют):
R-COOH + NaOH R-COO¯ Na+ + H2O
Однако по сравнению с минеральными кислотами карбоновые кислоты являются слабыми электролитами и их соли в водных растворах частично гидролизованы.
Оснóвные свойства карбоновых кислот проявляются у них по отношению к сильным кислотам. В смеси карбоновой кислоты с концентрированной серной кислотой или при растворении газообразного хлороводорода в карбоновой кислоте происходит её протонирование. Существование образующегося при этом катиона можно описать при помощи граничных структур:

Кислотно-основные свойства, кроме самих карбоновых кислот, присущи также амидам карбоновых кислот. В этих соединениях аминогруппа эффективно сопряжена с карбонильной:

При этом азот принимает sp2-гибридное состояние и вместе со своим окружением располагается в одной плоскости с карбонильной группой. В такой конформации и происходит эффективное р--сопряжение р-орбитали атома азота с -связью С=О.
В связи с р--сопряжением атом азота в амидах теряет оснóвные свойства, но при этом для амидов становятся более характерны кислотные свойства:

6.4.4.2. Реакции нуклеофильного замещения
Карбоновые кислоты и их ацильные производные аналогично альдегидам и кетонам могут подвергаться нуклеофильной атаке по карбонильному атому углерода. Однако эта реакция для них может быть затруднена из-за +М-эффекта заместителя Х, возникающего в результате р--сопряжения (см. выше). Поэтому реакционная способность этих соединений по отношению к нуклеофилам будет сильно зависеть от природы заместителя Х.
В отличие от альдегидов и кетонов карбоновые кислоты и их ацильные производные реагируют с нуклеофилами с образованием продуктов замещения, а не присоединения. Это связано с бóльшей устойчивостью продукта замещения по сравнению с промежуточной частицей и с устойчивостью аниона уходящей группы Х¯:

Нуклеофилами здесь обычно являются аммиак, амины, спирты, вода, их анионы, а также могут быть анионы карбоновых кислот, галогениды фосфора и серы. В целом для карбоновых кислот и их производных замещение протекает легче, чем нуклеофильное замещение у насыщенного атома углерода (в галогеналканах, спиртах, эфирах, аминах). Это связано, по крайней мере, с двумя причинами: 1) большей доступностью sp2-гибридизованного атома углерода в карбоксильной (или ацильной) группе по сравнению с тетраэдрическим в насыщенных соединениях; 2) более легким разрывом -связи в первом случае на лимитирующей стадии атаки реагентом по сравнению с разрывом-связи С–Х при SN2-механизме у sp3-гибридизованного атома углерода.
На скорость реакций ацилирования в большинстве случаев оказывают влияние кислотные катализаторы (протонные кислоты и кислоты Льюиса). При этом механизм катализа будет зависеть от типа кислоты:

или

Но производные карбоновых кислот могут сильно различаться по своей реакционной способности. На это влияют два фактора: величина донорного мезомерного эффекта заместителя X и устойчивость аниона уходящей группы Х¯.
В меньшей степени мезомерный эффект
проявляется для галогенов. Фтор наиболее
электроотрицателен, и донорный эффект
его незначителен.
Электроотрицательность от хлора к йоду
уменьшается хотя и значительно, но в
том же ряду сильно увеличиваются размеры
орбиталей внешнего уровня (на котором
находится неподелённая пара электронов),
поэтому взаимодействие такой орбитали
с C=О-связью
будет всё менее эффективным в этом ряду
галогенов. Напротив, наибольшим донорным
эффектом обладает аминогруппа, так как
азот менее электроотрицателен, чем фтор
и кислород, а внешние р-орбитали
имеют такую же геометрию, что и орбитали
-связи
C=О.
(Ещё больше +М-эффект
в анионах солей
меньшей степени мезомерный эффект
проявляется для галогенов. Фтор наиболее
электроотрицателен, и донорный эффект
его незначителен.
Электроотрицательность от хлора к йоду
уменьшается хотя и значительно, но в
том же ряду сильно увеличиваются размеры
орбиталей внешнего уровня (на котором
находится неподелённая пара электронов),
поэтому взаимодействие такой орбитали
с C=О-связью
будет всё менее эффективным в этом ряду
галогенов. Напротив, наибольшим донорным
эффектом обладает аминогруппа, так как
азот менее электроотрицателен, чем фтор
и кислород, а внешние р-орбитали
имеют такую же геометрию, что и орбитали
-связи
C=О.
(Ещё больше +М-эффект
в анионах солей
 ,
,
так как на частице делокализуется целочисленный отрицательный заряд. Это практически исключает взаимодействие с нуклеофилами таких частиц, и поэтому нуклеофильное замещение в анионах солей карбоновых кислот обычно не рассматривается.)
Устойчивость аниона уходящей группы для галогенов является наиболее высокой (галогенид-ионы — это анионы сильных кислот), а в противоположность им амид-ион малостабилен, так как является анионом слабой кислоты — аммиака.
Таким образом, все ацильные производные и сами кислоты можно расположить в следующий ряд уменьшения реакционной способности в реакциях нуклеофильного замещения: галогенангидриды, ангидриды, кислоты, сложные эфиры, амиды.
В этих реакциях замещения в преобладающем числе случаев одни ацильные производные превращаются в другие. Сравнивая их реакционную способность, можно предположить, что галогенангидриды будут легко превращаться как в сами кислоты, так и в другие их производные. И наоборот, амиды с большим трудом вступают в эти реакции: в жёстких условиях и в присутствии кислотного или основного катализатора.
Так, галогенангидриды могут превращаться в кислоты, ангидриды, сложные эфиры, амиды, а при действии кадмийорганических соединений — в кетоны:

Реакции идут легко, энергично, с большим тепловыделением. Применения катализатора не требуется. Последнее из приведённых превращений может использоваться в качестве одного из способов получения кетонов.
Механизм реакции, как и в случаях взаимодействия с нуклеофилами других ацильных производных, SN2. Например:

Однако для галогенангидридов (только!) возможен и SN1-механизм. На первой стадии образуется ацилий-катион, который дальше взаимодействует с нуклеофильной частицей:


Такой механизм наблюдается обычно в сильнокислой среде (концентрированная серная кислота, а также кислоты Льюиса), которая способствует диссоциации связи С–Х в молекулах галогенангидридов.
Ангидриды кислот — соединения тоже высокореакционноспособные и легко превращаются в кислоты и другие ацильные производные, кроме галогенангидридов, и по сравнению с галогенангидридами реагируют медленнее:

Реакция протекает по аналогичному механизму. Нуклеофильной атаке подвергается один из карбонильных атомов углерода — тот, который более доступен и на котором бóльший эффективный положительный заряд.
Сами карбоновые кислоты в большинство SN-реакций вступают в присутствии катализатора и/или при сильном нагревании. Так, для получения из кислот ангидридов обычно используется оксид фосфора (V) или оксид алюминия при нагревании, а галогенангидриды из кислот можно получить только при использовании таких мощных галогенирующих средств, как галогениды фосфора и серы (например, PCl5, SOCl2). Механизм реакции с тионилхлоридом возможно такой :


Общую схему превращения кислот в их производные можно представить следующим образом:

Механизм реакции образования из кислот сложных эфиров (т.е. этерификации), протекающей в кислой среде, можно представить следующим образом:


Такие ацильные производные, как сложные эфиры, ненамного отличаются по реакционной способности от карбоновых кислот, если реакцию проводят в присутствии кислотного катализатора. Кислотный гидролиз сложных эфиров является реакцией обратной этерификации, и все стадии его механизма осуществляются в обратном порядке:


Однако для третичных алкиловых эфиров (R= трет-алкил) механизм кислотного гидролиза может быть несколько иным:


В этом случае наблюдается разрыв связи O–Csp3, а механизм реакции можно классифицировать какSN1 у насыщенного атома углерода.
При оснóвном же катализе сложные эфиры даже более реакционноспособны, так как кислоты в щелочной среде превращаются в малоактивные карбоксилат-ионы, и реакция становится необратимой. К тому же нуклеофильной частицей здесь является более активный гидроксид-ион:


Общую схему превращений сложных эфиров по ацильной группе можно представить так:

Взаимодействие с молекулой другого спирта ROH и образование нового сложного эфира (это реакция переэтерификации) протекает по механизму, аналогичному гидролизу сложных эфиров. А взаимодействие с реактивами Гриньяра сначала даёт кетоны (как показано в схеме), а затем избыток реактива Гриньяра превращает кетоны в третичные спирты (гл. 6.1.4.1, п. 6). Образование кетонов — это тоже реакция нуклеофильного замещения, которое в этом случае протекает через образование четырёхцентровых переходного состояний:


Амиды карбоновых кислот, как уже отмечалось, труднее всего вступают в нуклеофильное замещение по ацильной группе, и единственной реакцией, имеющей практическое значение, здесь является гидролиз в кислой или щелочной среде с образованием карбоновой кислоты (или карбоксилат-иона). Например, для N,N-дизамещённых амидов:

Соли карбоновых кислот в реакции замещения по карбонильному атому углерода не вступают (см. выше строение карбоксилат-аниона), но в кислой среде они превращаются в карбоновые кислоты, и реакция замещения протекает как с карбоновыми кислотами:

6.4.4.3. Реакции с участием -водородных атомов
Одна из таких реакций — это бромирование (или иногда хлорирование) карбоновых кислот в присутствии красного фосфора (реакция Гелля–Фольгарда–Зелинского*), протекающее по схеме:
R–СН2–COОH
![]() R–СНBr–COОH
R–СНBr–COОH
Роль фосфора сводится к образованию на первой стадии трибромида фосфора, который, вступая в реакцию с молекулой кислоты, превращает её в галогенангидрид (см. выше):
P + Br2 PBr3

Образовавшийся бромоангидрид в енольной форме подвергается дальнейшему бромированию, а при взаимодействии со второй молекулой карбоновой кислоты происходит замещение в ацильной группе:


В результате образуется молекула -галогенозамещённой карбоновой кислоты и молекула галогенангидрида, которая дальше вступает в реакцию бромирования в енольной форме.
Использование избытка брома может привести к получению дибромозамещённых кислот:
R–СНBr–COОH
![]() R–СBr2–COОH
R–СBr2–COОH
Хлорирование так же, как и бромирование, протекает по электрофильному механизму, но возможно хлорирование и по радикальному механизму. В этом случае реакция будет проходить не только по -углеродному атому, например:

Помимо галогенирования для рассматриваемых соединений возможны и другие реакции с участием -водородных атомов. Эти реакции, вызванные проявлениемCН-кислотности данного структурного фрагмента, в бóльшей степени характерны для сложных эфиров, чем для каких-либо других ацильных производных кислот (так как кислоты и амиды обладают соответственноОН- и NН-кислотностью, а ангидриды и галогенангидриды высоко активны по отношению к нуклеофилам, роль которых могут играть участвующие в реакции основания). К таким реакциям относитсяконденсация Кляйзена*, в которую вступают две молекулы сложного эфира:
2
R-СН2-СООС2Н5
![]() R-СН2-СО-СНR-COОС2Н5
+
С2Н5ОН
R-СН2-СО-СНR-COОС2Н5
+
С2Н5ОН
Механизм реакции включает в себя отрыв протона от -углеродного атома под действием алкоголятов с образованием новой нуклеофильной частицы, которая атакует вторую молекулу сложного эфира:

Далее реакция развивается так же, как и при любом другом нуклеофильном замещении в ацильной группе. Заканчивается реакция отрывом алкоголят-аниона и образованием оксоэфира:

К реакциям с участием -водородных атомов можно отнести также образование кетенов при действии сильных органических оснований на галогенангидриды кислот. При этом протекает реакция отщепления и замыкается-связь (гл. 6.2.1).
6.4.4.4. Декарбоксилирование кислот и их солей
Декарбоксилирование может наблюдаться при сплавлении солей карбоновых кислот со щелочами (при 500—600 К). Эта реакция используется для получения низших алканов. Здесь в условиях сильного нагревания происходит нуклеофильное замещение алкильного радикала:

Пиролиз бариевых или кальциевых солей карбоновых кислот также приводит к декарбоксилированию и служит одним из лабораторных способов получения кетонов:

Декарбоксилирование может наблюдаться и при электролизе натриевых и калиевых солей карбоновых кислот (реакция Кольбе):
|
(на аноде) |
|
![]()
При этом образуются алканы с чётным числом атомов углерода в молекуле, и поэтому электролиз является одним из лабораторных способов получения предельных углеводородов (гл. 2.5).
По радикальному механизму также происходит декарбоксилирование серебряных солей карбоновых кислот при обработке их хлором или бромом (реакция Хунсдиккера–Бородина*):

![]()
6.4.4.5. Перегруппировка амидов по Гофману
Эта реакция является одним из способов получения чистых первичных аминов: амид при действии на него брома (или хлора) в щелочной среде превращается в амин с уменьшением углеродной цепи на один атом. Механизм можно представить следующим образом:


(Отщепление бромид-иона и карбанионное перемещение происходят синхронно.)
Образовавшийся в результате перегруппировки алкилизоцианат затем гидролизуется:


6.4.4.6. Дегидратация амидов
Это ещё одно свойство амидов, связанное с большой подвижностью атомов водорода при азоте. В результате под действием фосфорного ангидрида происходит отщепление молекулы воды и образование другого производного кислоты — нитрила:

6.4.4.7. Нуклеофильное присоединение к нитрилам
Нитрилы карбоновых кислот вследствие строения их функциональной группы (гл. 6.4.2) должны вступать в реакции нуклеофильного присоединения (AdN) аналогично альдегидам. Среди наиболее распространённых примеров — реакции присоединения воды, спиртов, хлороводорода с образованием соответственно амидов кислот, иминоэфиров и хлориминов:

Присоединение воды возможно как в кислой, так и в щелочной среде. Механизм реакции:



Спирты присоединяются в кислой среде в отсутствие воды. Механизм реакции:
