

§ 4. Количественное определение оксида углерода (II) в крови
![]()
![]()
![]()
![]() Все
объявления
Все
объявления
ЯндексДирект
Дать объявление
Низкотемпературные камеры
низкотемпературные морозильники с температурным режимом -24С, -55С, -85С
Адрес и телефон· www.winecoolers.ru
![]() Содержаниеоксида
углерода(II) в крови
определяют по количеству карбоксигемоглобина.
Для этой цели может быть использован
спектрофотометрический метод, предложенный
В. Ф. Крамаренко, Б. А. Собчуком и Т. Н.
Гладышевской, который приведен в
«Методических указаниях о количественном
определении карбоксигемоглобина и
карбоксимиоглобина» (1974).
Содержаниеоксида
углерода(II) в крови
определяют по количеству карбоксигемоглобина.
Для этой цели может быть использован
спектрофотометрический метод, предложенный
В. Ф. Крамаренко, Б. А. Собчуком и Т. Н.
Гладышевской, который приведен в
«Методических указаниях о количественном
определении карбоксигемоглобина и
карбоксимиоглобина» (1974).
Поступивший в организмоксид углерода(II) связывается с дезокси- иоксигемоглобином, вследствие чего образуется карбоксигемоглобин (COHb).Метгемоглобинне связывается соксидом углерода(II) в крови. Однако в лабораторных условиях при помощи дитионитанатрия(Na2S2O4·2H2O) или другихвосстановителейметгемоглобинможно перевести в дезоксигемоглобин.
В ряде источников литературы дитионит натриявстречается под названием «гидросульфит натрия».
Все перечисленные выше соединения гемоглобина(дезоксигемоглобин,оксигемоглобини карбоксигемоглобин) можно обнаружить по их спектрам поглощения в видимой области в пределах длин волн от 450 до 620 нм. Спектры поглощенияоксигемоглобинаи карбоксигемоглобина незначительно отличаются друг от друга. В связи с этим спектральные характеристики указанных соединений трудно использовать для их количественного определения. Значительно отличаются друг от друга спектры поглощения дезоксигемоглобина и карбоксигемоглобина. Поэтому различие этих спектров используется для количественного определения карбоксигемоглобина в крови.
Для количественного спектрофотометрического определения оксида углерода(II) по карбоксигемоглобину приготовляют рядрастворов:растворА —растворисследуемой крови;растворБ —растворкрови, содержащей смесь карбоксигемоглобина и дезоксигемоглобина;растворВ —растворкрови, в которой все формыгемоглобина(дезоксигемоглобин,оксигемоглобини метгемоглобин) полностью переведены в карбоксигемоглобин.
Чтобы избежать частичного разложения карбоксигемоглобина работа с содержащими его растворамидолжна производиться вдали от естественных и искусственных источников света с минимальным доступомвоздуха.
Приготовление раствора А.1 мл исследуемой трупной крови, не содержащей сгустков, вносят в мерную колбу вместимостью 100 мл и прибавляют фосфатныйбуферный раствор(рН = 7,38).Жидкостьвзбалтывают и объем ее доводят фосфатнымбуферным растворомдо 100 мл. Полученный при этомрастворкрови должен быть прозрачным.
Приготовление раствора Б.Этотрастворготовят израствораА непосредственно в кювете спектрофотометра перед измерением оптической плотности. С этой целью в кювету вносятрастворисследуемой крови (раствор А) в таком количестве, чтобы после закрытия кюветы крышкой между ней ижидкостьюне быловоздуха. КрастворуА, внесенному в кювету, прибавляют 3—4 мг дитионитанатрия(Na2S2O4·H2O). Содержимое кюветы тщательно перемешивают тонкой стекляннойпалочкой. При этомоксигемоглобиниметгемоглобинвосстанавливаются до дезоксигемоглобина, а карбоксигемоглобин с дитионитомнатрияне реагирует. После прибавления дитионитанатрияраствордолжен быть прозрачным.
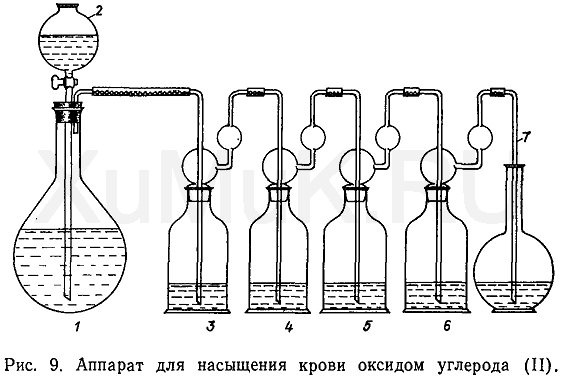
Приготовление раствора В.Этотрастворполучают в специальном приборе, представленном на рис. 9. Применяемый для этой цели прибор состоит из колбы 1, закрытой пробкой, снабженной капельной воронкой 2 и отводной стеклянной трубкой для выходаоксида углерода(II) из колбы, четырех склянок Дрек-селя (3, 4, 5, 6) и отводной трубки 7. Колбу и склянки Дрекселя соединяют между собой резиновыми трубками. При отсутствии склянок Дрекселя их можно заменить колбами вместимостью 50 мл, отверстия которых закрыты пробками, снабженными двумя стеклянными трубками.
В колбу 1 вносят 50 мл концентрированной серной кислоты, а в капельную воронку 2 — 10 млмуравьиной кислоты. В склянку 3 вносят 10 %-йрастворгидроксида натрия, в склянки 4 и 6 —дистиллированную воду, а в склянку 5 —растворА исследуемой крови в фосфатной буферной смеси. В склянки 3, 4, 5 и 6 вносят столькожидкости, чтобы трубки погружались на 2 см вжидкость.
Из капельной воронки 2 в подогретую колбу / по каплям приливают муравьиную кислоту. Интенсивность выделенияоксида углерода(II) регулируют скоростью приливаниямуравьиной кислоты. По мере расходованиямуравьиной кислотывыделениегазазамедляется. В начале опыта для увеличения скорости выделенияоксида углерода(II) колбу осторожно нагревают на небольшом пламени газовой горелки.
Учитывая высокую токсичностьоксида углерода(II), при работе с ним необходимо соблюдать осторожность. Получениеоксида углерода(II) и насыщение крови этимгазомдолжно производиться в вытяжном шкафу с хорошей тягой.
Оксид углерода(II) из колбы 1 пропускают через склянки Дрекселя в течение 15 мин. За это времяоксигемоглобинкрови полностью превращается в карбоксигемоглобин. Однако при этом враствореможет оставаться некоторое количествометгемоглобина, который необходимо перевести в дезоксигемоглобин, а затем в карбоксигемоглобин. С указанной целью после пятиминутного пропусканияоксида углерода(II) от прибора отсоединяют склянку 5, в которую вносят 5—7 мг дитионитанатрия, ижидкостьхорошо взбалтывают. (Осторожно! Не вдыхатьоксид углерода(II)!). Затем склянку 5 присоединяют к прибору и в течение 5 мин пропускаютоксид углерода(II). После насыщенияоксидом углерода(II)растворкрови, содержащий карбоксигемоглобин, должен быть прозрачным.
При количественном определении оксида углерода(II) необходимо измерять оптическую плотностьрастворакрови, содержащего смесь карбоксигемоглобина и дезоксигемоглобина, а затем измерять оптическую плотностьрастворакрови, насыщенногооксидом углерода(II). Этотрастворне должен содержать дезоксигемоглобина,оксигемоглобинаиметгемоглобина.
Дезоксигемоглобин имеет максимум светопоглощения при длине волны, равной 557 нм, а карбоксигемоглобин имеет 2 максимума светопоглощения при длинах волн 541 и 571 нм. При наложении спектральных кривых карбоксигемоглобина и дезоксигемоглобина на одном графике отмечается появление трех изобестических точек (а, б, в) при длинах волн 550, 565 и 580 нм. В этих точках пересечения спектральных кривых оптические плотности растворовкарбоксигемоглобина и дезоксигемоглобина одинаковы (рис. 10).

Прежде чем приступить к определению карбоксигемоглобина спектрофотометрическим методом, на графике, на который нанесены спектры поглощения карбоксигемоглобина и дезоксигемоглобина, необходимо найти длину волны, при которой расстояние между обеими спектральными кривыми (карбоксигемоглобина и дезоксигемоглобина) будет наибольшим вблизи первого максимума поглощения карбоксигемоглобина (т. е. при длине волны, равной 541 нм). На основании экспериментальных данныхэта наибольшая разница значений оптических плотностейрастворовкарбоксигемоглобина и дезоксигемоглобина имеет место при длине волны, равной 538 нм.
В кювету спектрофотометра с толщиной слоя жидкости1 см вносят исследуемыйрастворкрови (раствор А), прибавляют 3— 4 мг дитионитанатрияи поступают так, как указано при описании способа полученияраствораБ. Оптическую плотность этогораствораизмеряют при длинах волн, равных 538 и 550 нм. Затем измеряют оптическую плотностьрастворакрови, в которой весьгемоглобинпереведен в карбоксигемоглобин (раствор В) при длине волны, равной 538 нм. При измерении оптической плотности обоихрастворовкровирастворомсравнения являетсявода.
Приготовление сульфида аммония (см. Приложение 1, реактив 73).
Приготовление фосфатного буферного раствора для определения карбоксигемоглобина (см. Приложение 1, реактив 74).
Расчет содержания карбоксигемоглобина в исследуемой крови в процентах Ρ производят по формуле

где D COHb— оптическая плотностьраствораВ крови, дополнительно насыщенногооксидом углерода(II) (при 538 нм); DHbCOHb— оптическая плотностьраствораБ крови, обработанного дитионитомнатрия, содержащего смесь дезокси- и карбоксигемоглобина (при 538 нм); DHbI— оптическая плотностьраствораБ крови в изобестической точке (при 550 нм); К — коэффициент 0,372.
Величина ошибки определения карбоксигемоглобина в пределах концентрацийот 3 до 20 % составляет ±3%, приконцентрацияхсвыше 20 % погрешность примерно равняется ± 5 %.
Вариант метода спектрофотометрического определения карбоксигемоглобина в крови предложили Л. П. Букина и Л. И. Ушакова (Судебно-медицинская экспертиза, 1979, № 2). Предложенный ими вариант метода аналогичен описанному выше. Однако, согласно варианту метода, предложенного Л. П. Букиной и Л. И. Ушаковой, не требуется насыщения крови оксидом углерода(II) при каждом определении карбоксигемоглобина.
Судебно-медицинская оценка результатов количественного определения карбоксигемоглобина в крови по Г. А. Сыцянко (Научно-исследовательский институт судебной медицины МЗ СССР) приведена ниже.
Содержание карбоксигемоглобина в крови зависит прежде всего от концентрацииоксида углерода(II) во вдыхаемомвоздухеи времени его воздействия.Концентрациякарбоксигемоглобина в крови тем выше, чем выше парциальноедавлениеоксида углерода(II) в альвеолярномвоздухепо сравнению с парциальнымдавлениемкислорода.
За один и тот же промежуток времени при прочих равных условиях в организмпоступаетоксида углерода(II) тем больше, чем больше минутный объемдыхания. Симптомы, обусловленные разной коицетрацией карбоксигемоглобина в крови, тяжесть и исход отравления представлены ниже в табл. 9. Этиданныеимеют ориентировочное значение.
Однако наблюдения и специальные исследования показывают, что соответствие между концентрациейкарбоксигемоглобина и тяжестью отравления имеется не всегда. Это особенно отчетливо проявляется при групповых отравлениях.
Смертельная концентрациякарбоксигемоглобина в крови составляет в среднем около 60 %, но может колебаться от 40 до 80 % и более. Это колебание обусловлено как влиянием внешних условий, так и особенностямиорганизма.

Более подробно судебно-медицинская оценка результатов количественного определения карбоксигемоглобина приведена в упомянутых выше «Методических указаниях о количественном определении карбоксигемоглобина и карбоксимиоглобина».